

From cells to tissue: How cell scale heterogeneity impacts glioblastoma growth and treatment response
- PMID: 32101537
- PMCID: PMC7062288
- DOI: 10.1371/journal.pcbi.1007672
From cells to tissue: How cell scale heterogeneity impacts glioblastoma growth and treatment response
Abstract
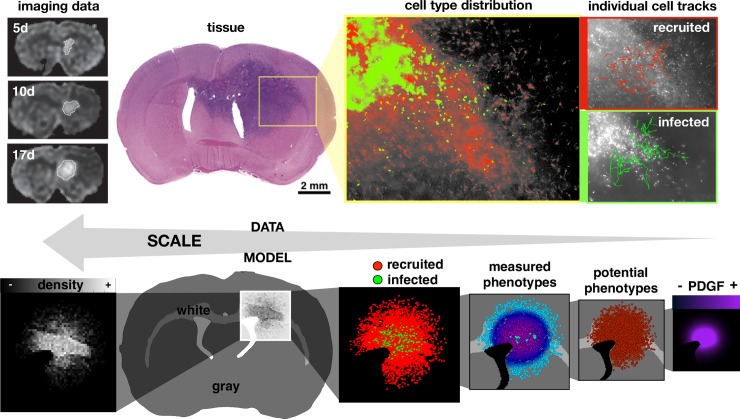
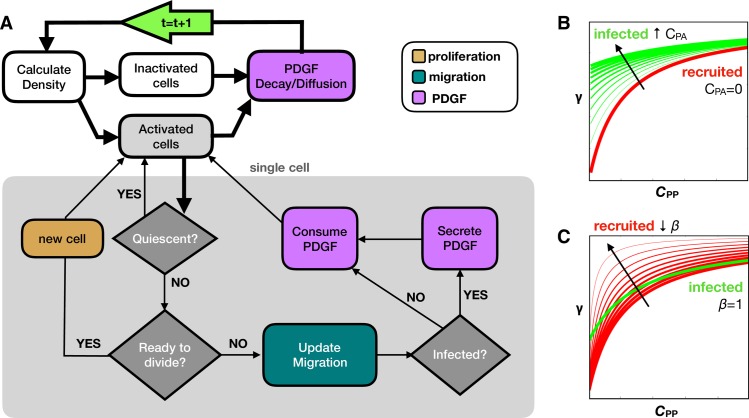
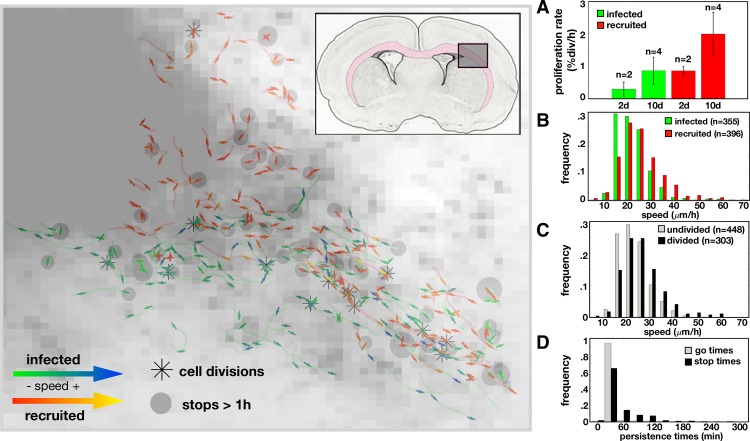
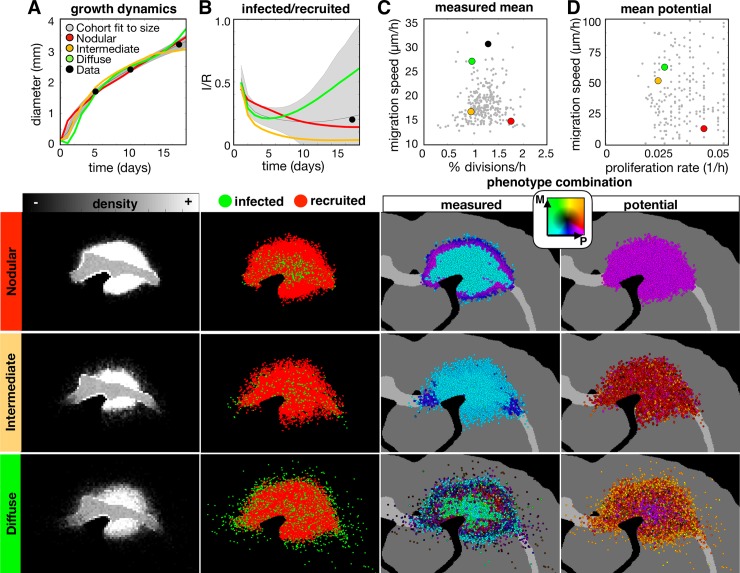
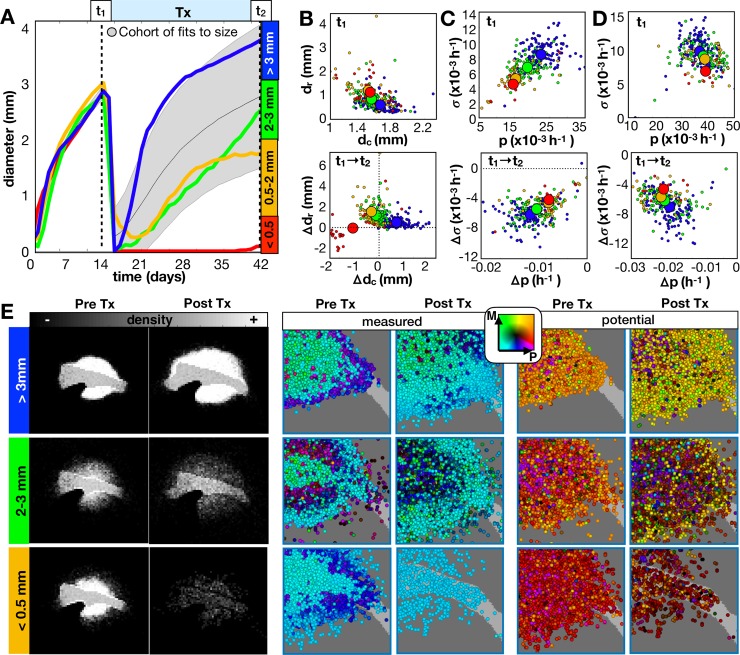
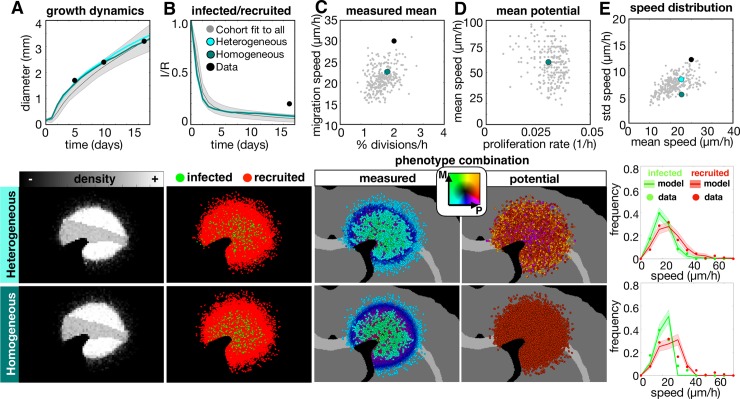
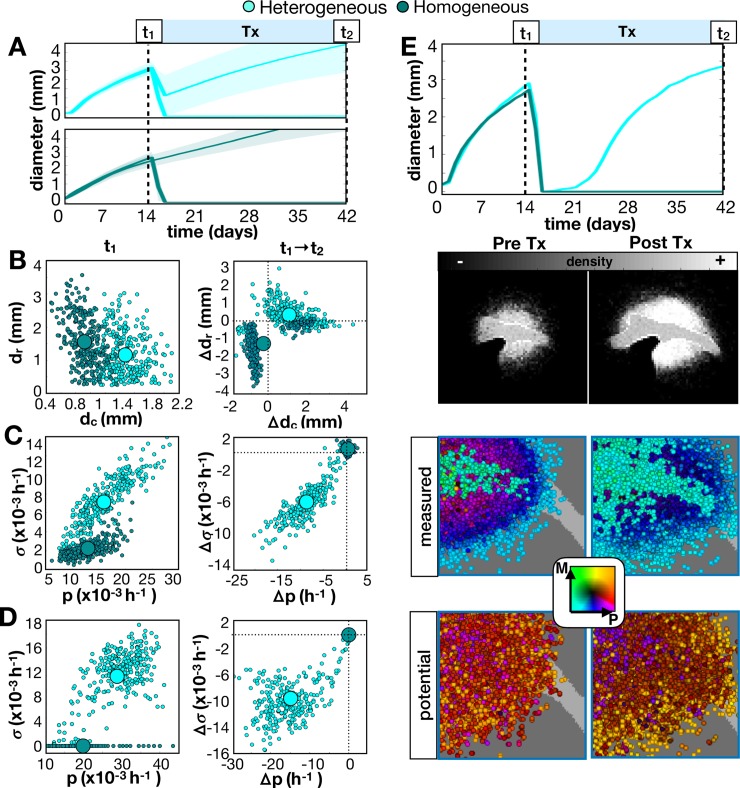
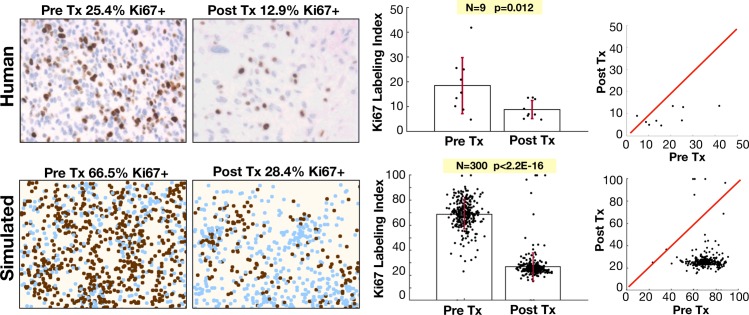
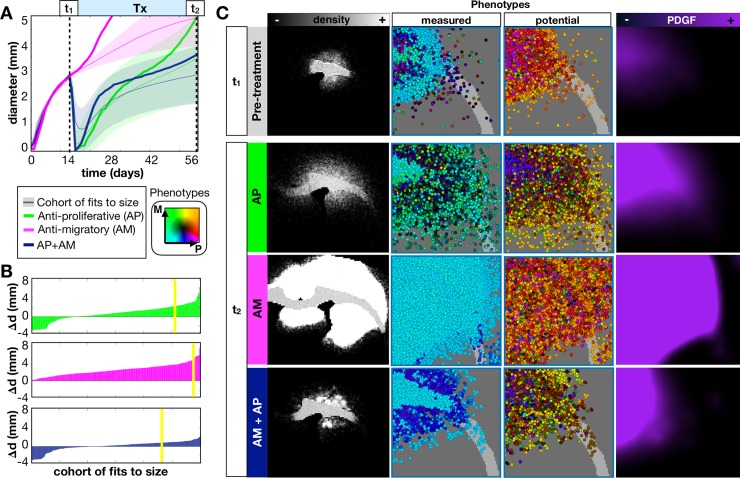
Glioblastomas are aggressive primary brain tumors known for their inter- and intratumor heterogeneity. This disease is uniformly fatal, with intratumor heterogeneity the major reason for treatment failure and recurrence. Just like the nature vs nurture debate, heterogeneity can arise from intrinsic or environmental influences. Whilst it is impossible to clinically separate observed behavior of cells from their environmental context, using a mathematical framework combined with multiscale data gives us insight into the relative roles of variation from different sources. To better understand the implications of intratumor heterogeneity on therapeutic outcomes, we created a hybrid agent-based mathematical model that captures both the overall tumor kinetics and the individual cellular behavior. We track single cells as agents, cell density on a coarser scale, and growth factor diffusion and dynamics on a finer scale over time and space. Our model parameters were fit utilizing serial MRI imaging and cell tracking data from ex vivo tissue slices acquired from a growth-factor driven glioblastoma murine model. When fitting our model to serial imaging only, there was a spectrum of equally-good parameter fits corresponding to a wide range of phenotypic behaviors. When fitting our model using imaging and cell scale data, we determined that environmental heterogeneity alone is insufficient to match the single cell data, and intrinsic heterogeneity is required to fully capture the migration behavior. The wide spectrum of in silico tumors also had a wide variety of responses to an application of an anti-proliferative treatment. Recurrent tumors were generally less proliferative than pre-treatment tumors as measured via the model simulations and validated from human GBM patient histology. Further, we found that all tumors continued to grow with an anti-migratory treatment alone, but the anti-proliferative/anti-migratory combination generally showed improvement over an anti-proliferative treatment alone. Together our results emphasize the need to better understand the underlying phenotypes and tumor heterogeneity present in a tumor when designing therapeutic regimens.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Saeed-Vafa D, Bravo R, Dean JA, El-Kenawi A, Mon Père N, Strobl M, et al. Combining radiomics and mathematical modeling to elucidate mechanisms of resistance to immune checkpoint blockade in non-small cell lung cancer. Bioarxiv. 2017;190561.
