

Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends
- PMID: 32101727
- PMCID: PMC7199893
- DOI: 10.1016/j.trecan.2019.12.009
Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends
Abstract
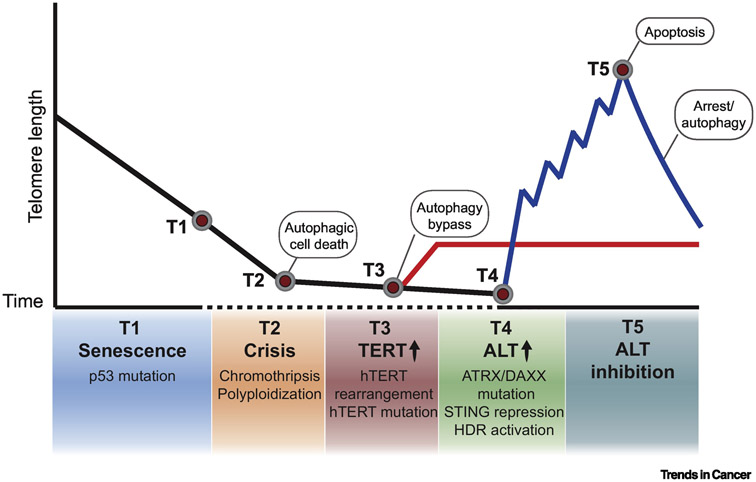
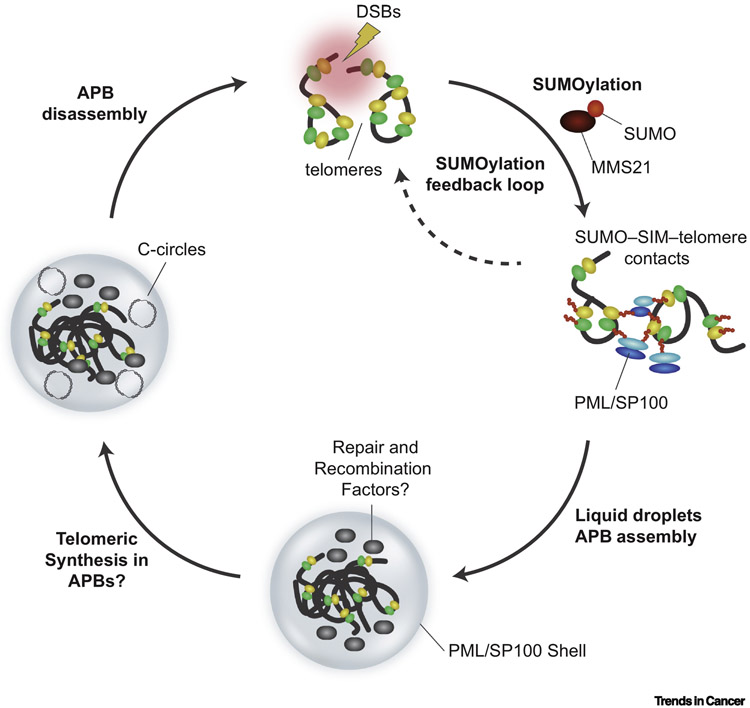
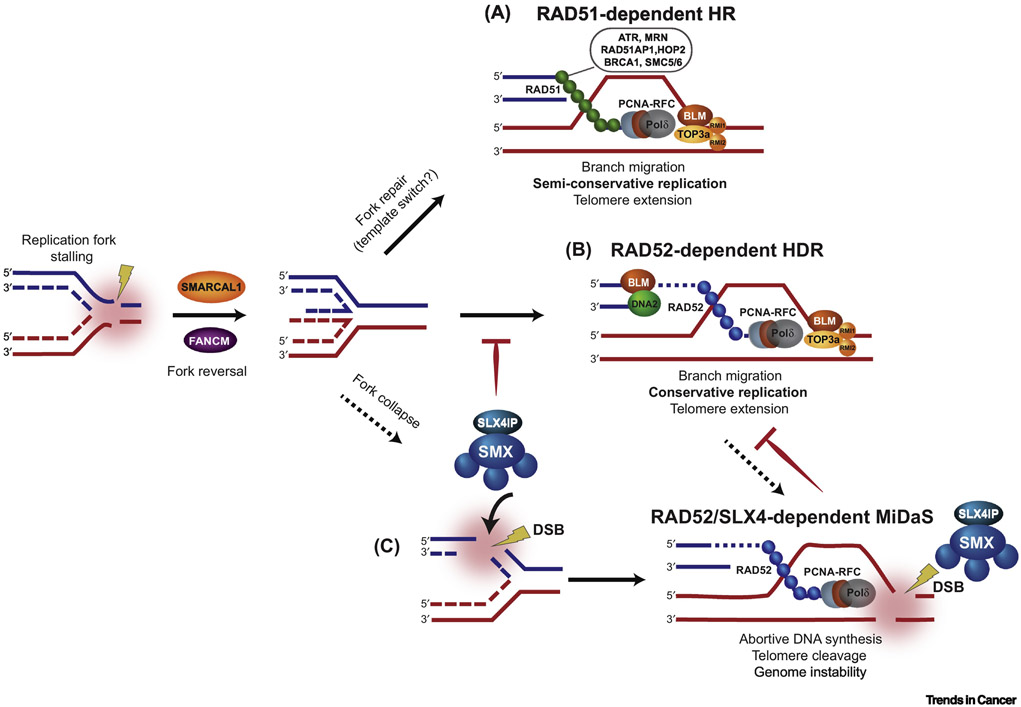
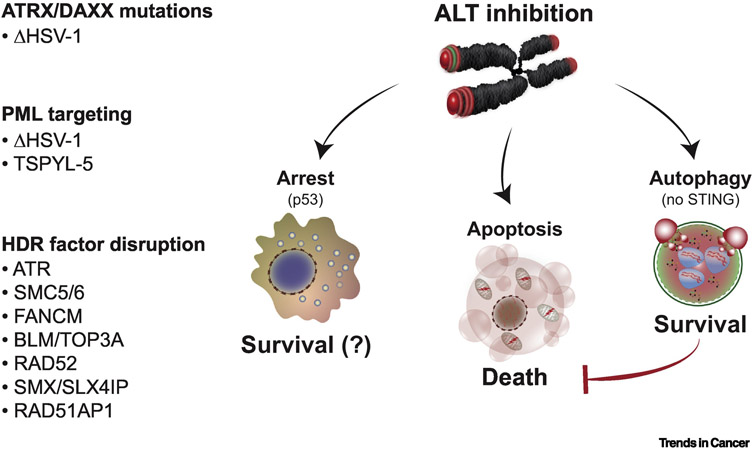
Alternative lengthening of telomeres (ALT) is a mechanism of telomere maintenance that is observed in many of the most recalcitrant cancer subtypes. Telomeres in ALT cancer cells exhibit a distinctive nucleoprotein architecture shaped by the mismanagement of chromatin that fosters cycles of DNA damage and replicative stress that activate homology-directed repair (HDR). Mutations in specific chromatin-remodeling factors appear to be key determinants of the emergence and survival of ALT cancer cells. However, these may represent vulnerabilities for the targeted elimination of ALT cancer cells that infiltrate tissues and organs to become devastating tumors. In this review we examine recent findings that provide new insights into the factors and mechanisms that mediate telomere length maintenance and survival of ALT cancer cells.
Keywords: DNA damage; cancer; homology-directed repair; replicative stress; telomere.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Figures
Similar articles
-
Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway.PLoS Genet. 2012;8(7):e1002772. doi: 10.1371/journal.pgen.1002772. Epub 2012 Jul 19. PLoS Genet. 2012. PMID: 22829774 Free PMC article.
-
Alternative Lengthening of Telomeres: DNA Repair Pathways Converge.Trends Genet. 2017 Dec;33(12):921-932. doi: 10.1016/j.tig.2017.09.003. Epub 2017 Sep 29. Trends Genet. 2017. PMID: 28969871 Review.
-
Life and cancer without telomerase: ALT and other strategies for making sure ends (don't) meet.Crit Rev Biochem Mol Biol. 2017 Feb;52(1):57-73. doi: 10.1080/10409238.2016.1260090. Epub 2016 Nov 28. Crit Rev Biochem Mol Biol. 2017. PMID: 27892716 Free PMC article. Review.
-
The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening.Nat Struct Mol Biol. 2019 Mar;26(3):213-219. doi: 10.1038/s41594-019-0192-3. Epub 2019 Mar 4. Nat Struct Mol Biol. 2019. PMID: 30833786 Free PMC article.
-
Anti-recombination function of MutSα restricts telomere extension by ALT-associated homology-directed repair.Cell Rep. 2021 Dec 7;37(10):110088. doi: 10.1016/j.celrep.2021.110088. Cell Rep. 2021. PMID: 34879271 Free PMC article.
Cited by
-
Mutational spectrum of ATRX aberrations in neuroblastoma and associated patient and tumor characteristics.Cancer Sci. 2022 Jun;113(6):2167-2178. doi: 10.1111/cas.15363. Epub 2022 Apr 26. Cancer Sci. 2022. PMID: 35384159 Free PMC article.
-
Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution.Biology (Basel). 2023 Apr 28;12(5):671. doi: 10.3390/biology12050671. Biology (Basel). 2023. PMID: 37237485 Free PMC article. Review.
-
TGF-β superfamily-induced transcriptional activation pathways establish the RAD52-dependent ALT machinery during malignant transformation of MPNSTs.Sci Rep. 2024 Nov 2;14(1):26475. doi: 10.1038/s41598-024-76732-z. Sci Rep. 2024. PMID: 39488637 Free PMC article.
-
Using telomeric chromosomal aberrations to evaluate clastogen-induced genomic instability in mammalian cells.Chromosome Res. 2020 Dec;28(3-4):259-276. doi: 10.1007/s10577-020-09641-2. Epub 2020 Sep 17. Chromosome Res. 2020. PMID: 32940874 Review.
-
Potential clinical treatment prospects behind the molecular mechanism of alternative lengthening of telomeres (ALT).J Cancer. 2023 Jan 31;14(3):417-433. doi: 10.7150/jca.80097. eCollection 2023. J Cancer. 2023. PMID: 36860927 Free PMC article. Review.
References
-
- de Lange T (2018) Shelterin-Mediated Telomere Protection. Anmi. Rev. Genet 52, 223–247 - PubMed
-
- Griffith JD et al. (1999) Mammalian telomeres end in a large duplex loop. Cell 97, 503–514 - PubMed
-
- Van Ly D et al. (2018) Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 71, 510–525.e6 - PubMed
-
- Denchi EL and de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
