

Dissecting the Regulatory Strategies of NF-κB RelA Target Genes in the Inflammatory Response Reveals Differential Transactivation Logics
- PMID: 32101750
- PMCID: PMC7061728
- DOI: 10.1016/j.celrep.2020.01.108
Dissecting the Regulatory Strategies of NF-κB RelA Target Genes in the Inflammatory Response Reveals Differential Transactivation Logics
Abstract
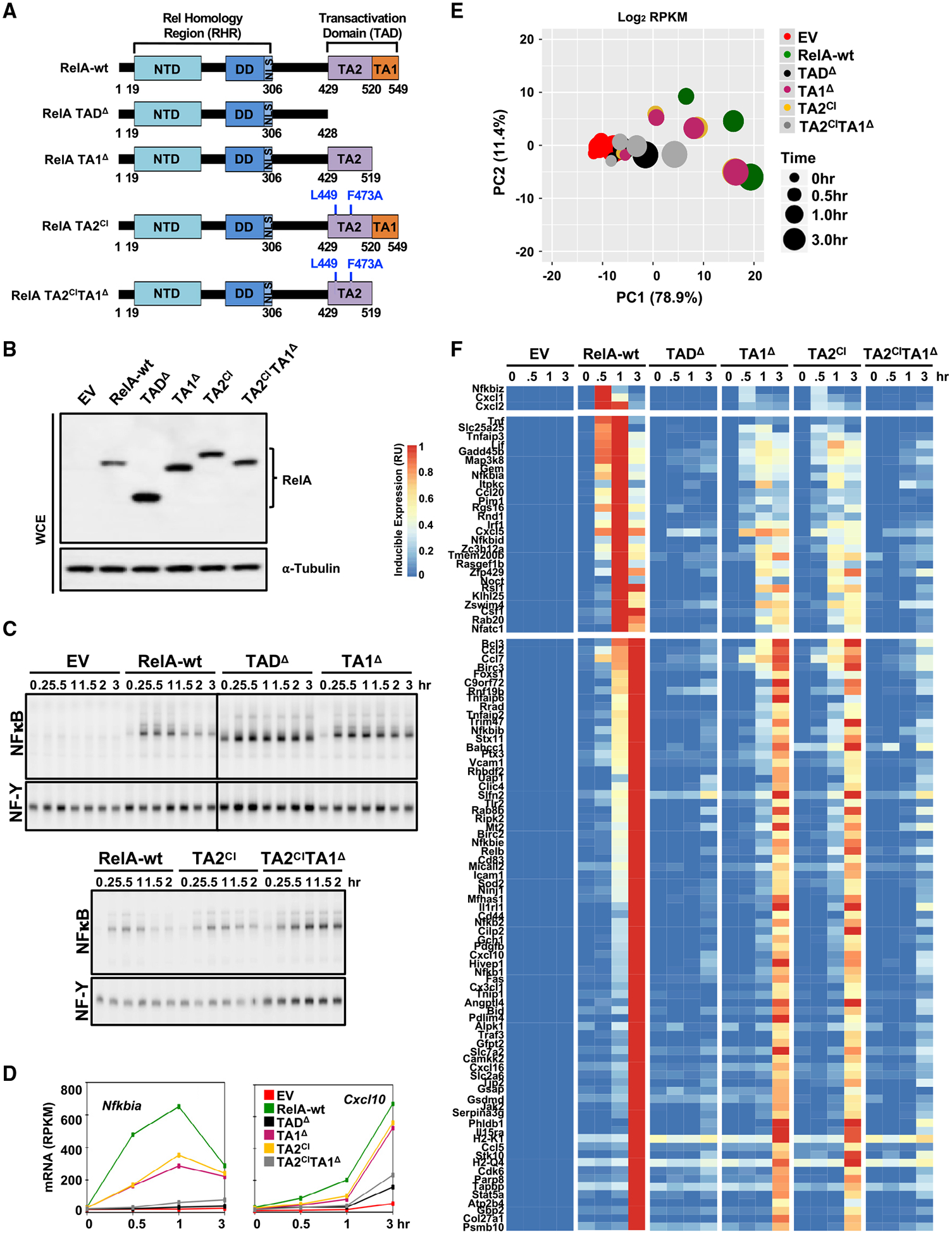
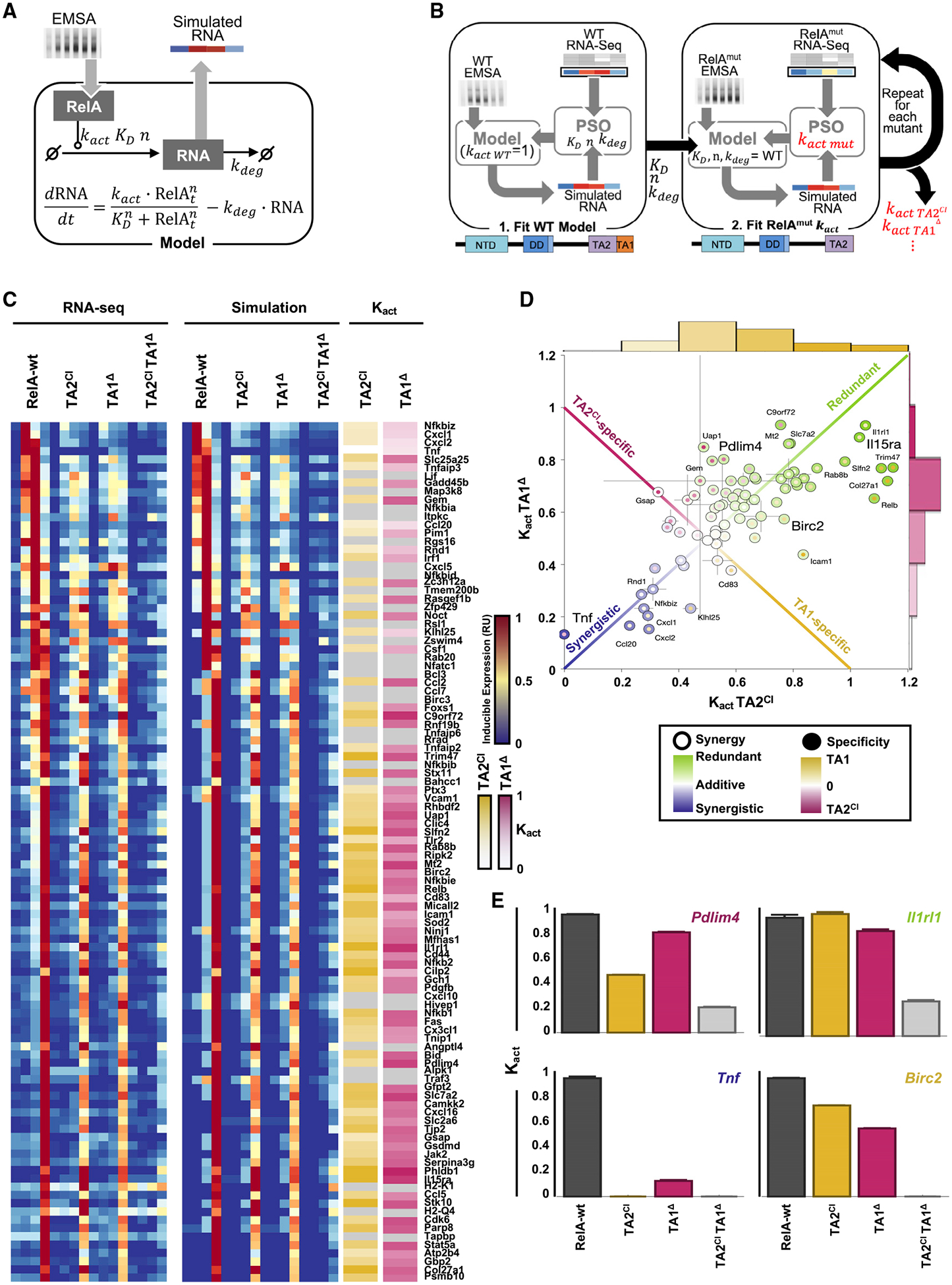
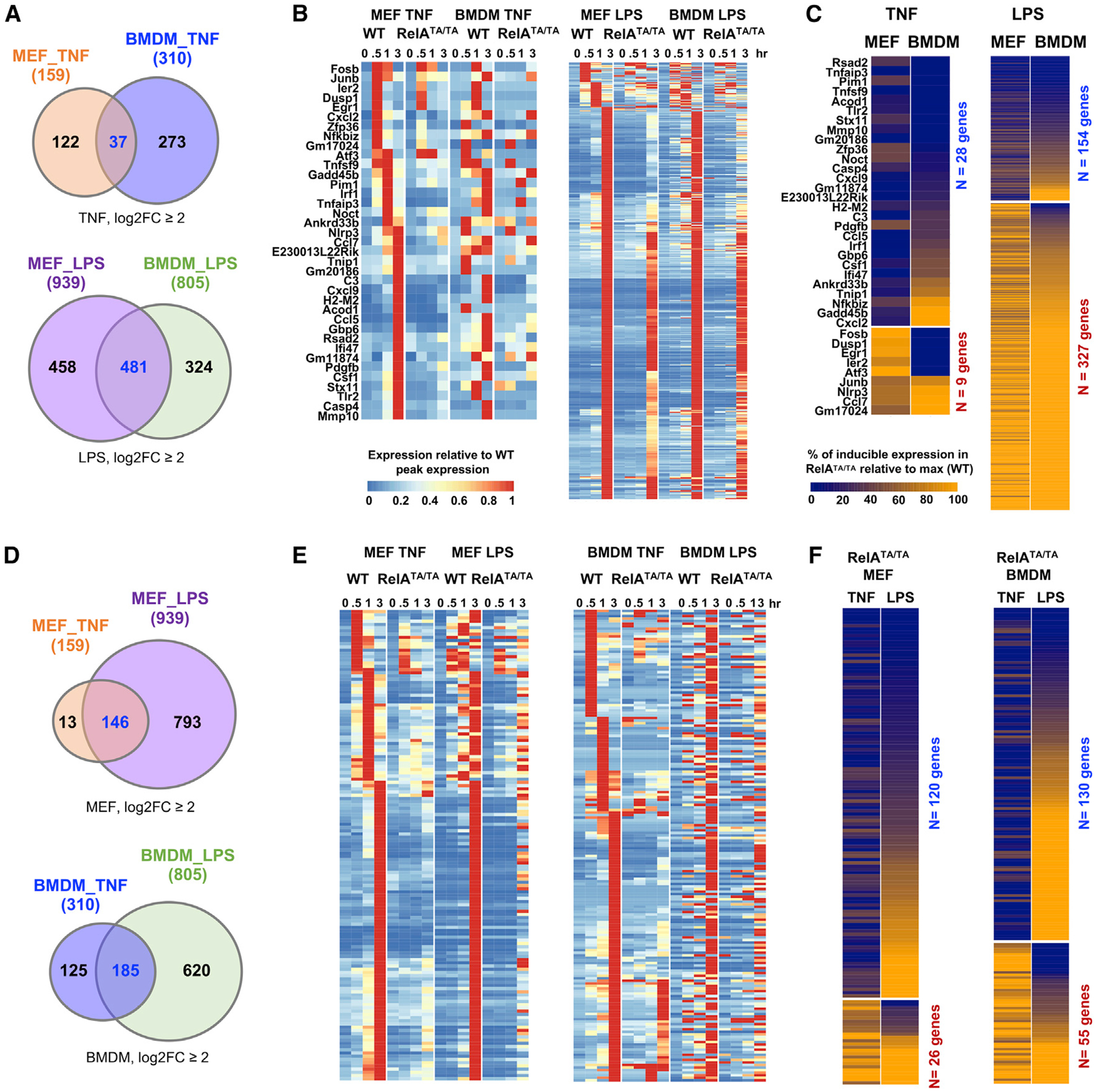
Nuclear factor κB (NF-κB) RelA is the potent transcriptional activator of inflammatory response genes. We stringently defined a list of direct RelA target genes by integrating physical (chromatin immunoprecipitation sequencing [ChIP-seq]) and functional (RNA sequencing [RNA-seq] in knockouts) datasets. We then dissected each gene's regulatory strategy by testing RelA variants in a primary-cell genetic-complementation assay. All endogenous target genes require RelA to make DNA-base-specific contacts, and none are activatable by the DNA binding domain alone. However, endogenous target genes differ widely in how they employ the two transactivation domains. Through model-aided analysis of the dynamic time-course data, we reveal the gene-specific synergy and redundancy of TA1 and TA2. Given that post-translational modifications control TA1 activity and intrinsic affinity for coactivators determines TA2 activity, the differential TA logics suggests context-dependent versus context-independent control of endogenous RelA-target genes. Although some inflammatory initiators appear to require co-stimulatory TA1 activation, inflammatory resolvers are a part of the NF-κB RelA core response.
Keywords: NF-kappaB; NF-κB; gene regulatory strategies; immune response; inflammatory response; mathematical modeling; structure-function analysis; systems biology; transcriptional activation; transcriptional synergy.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
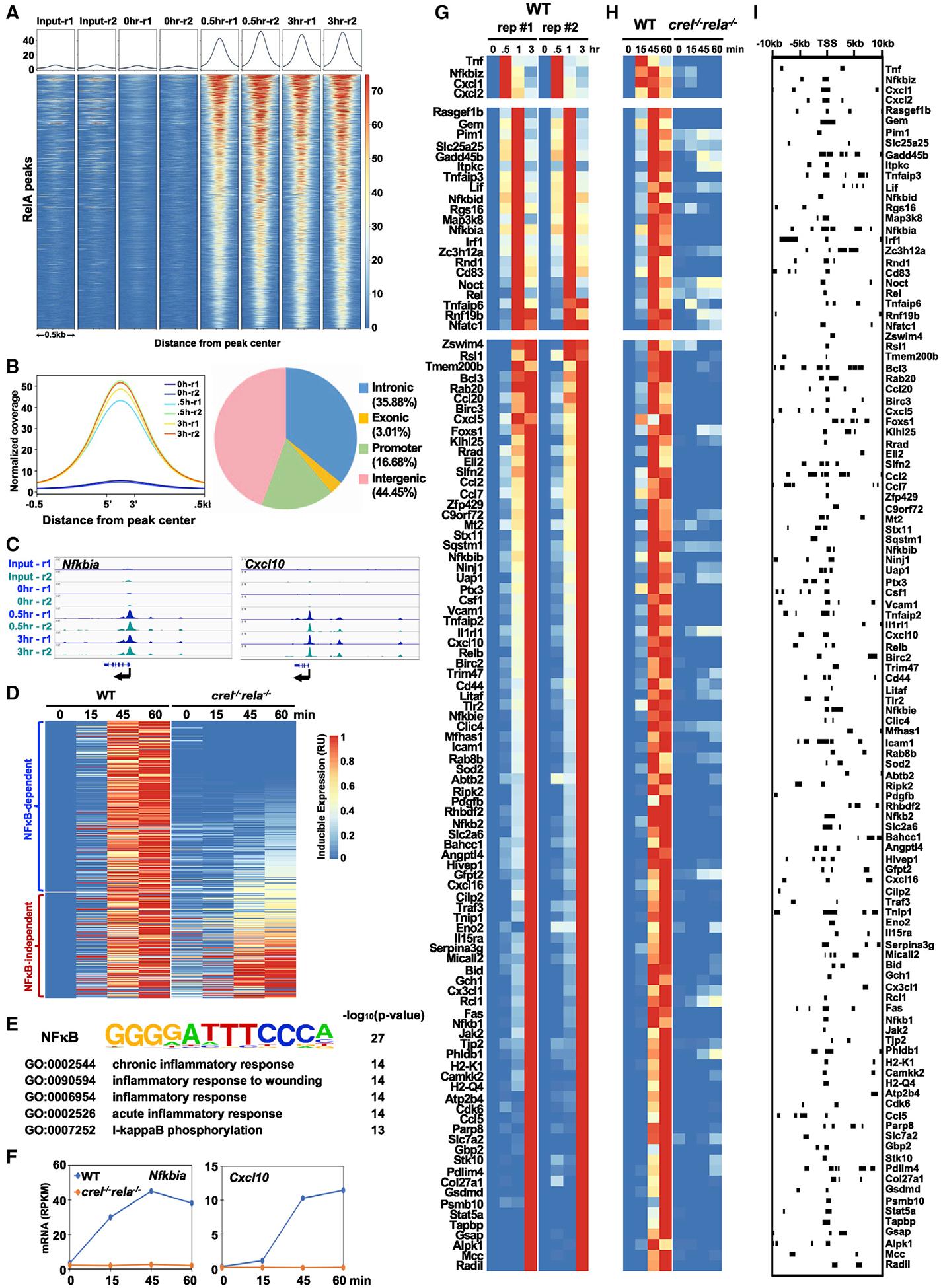
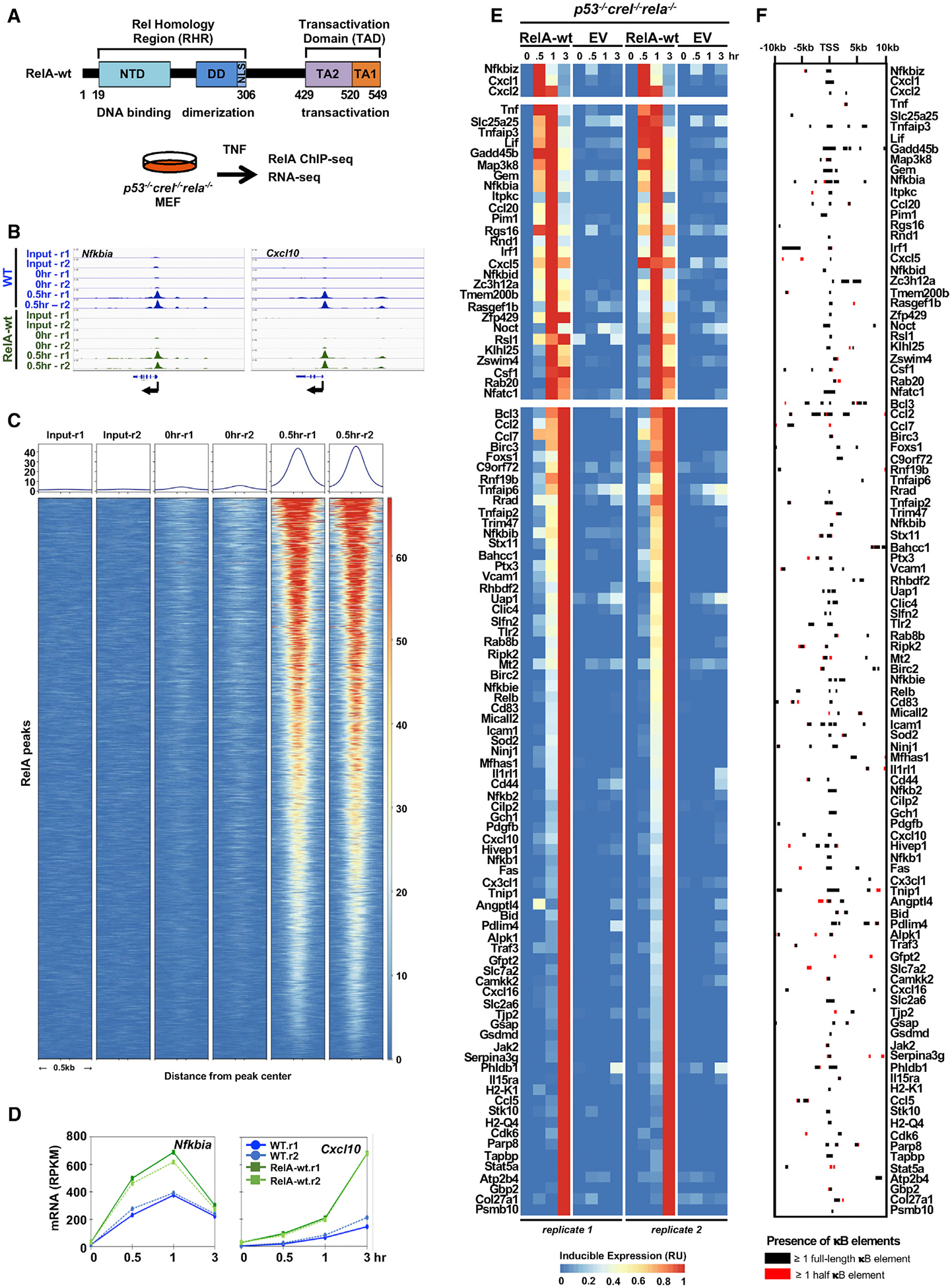
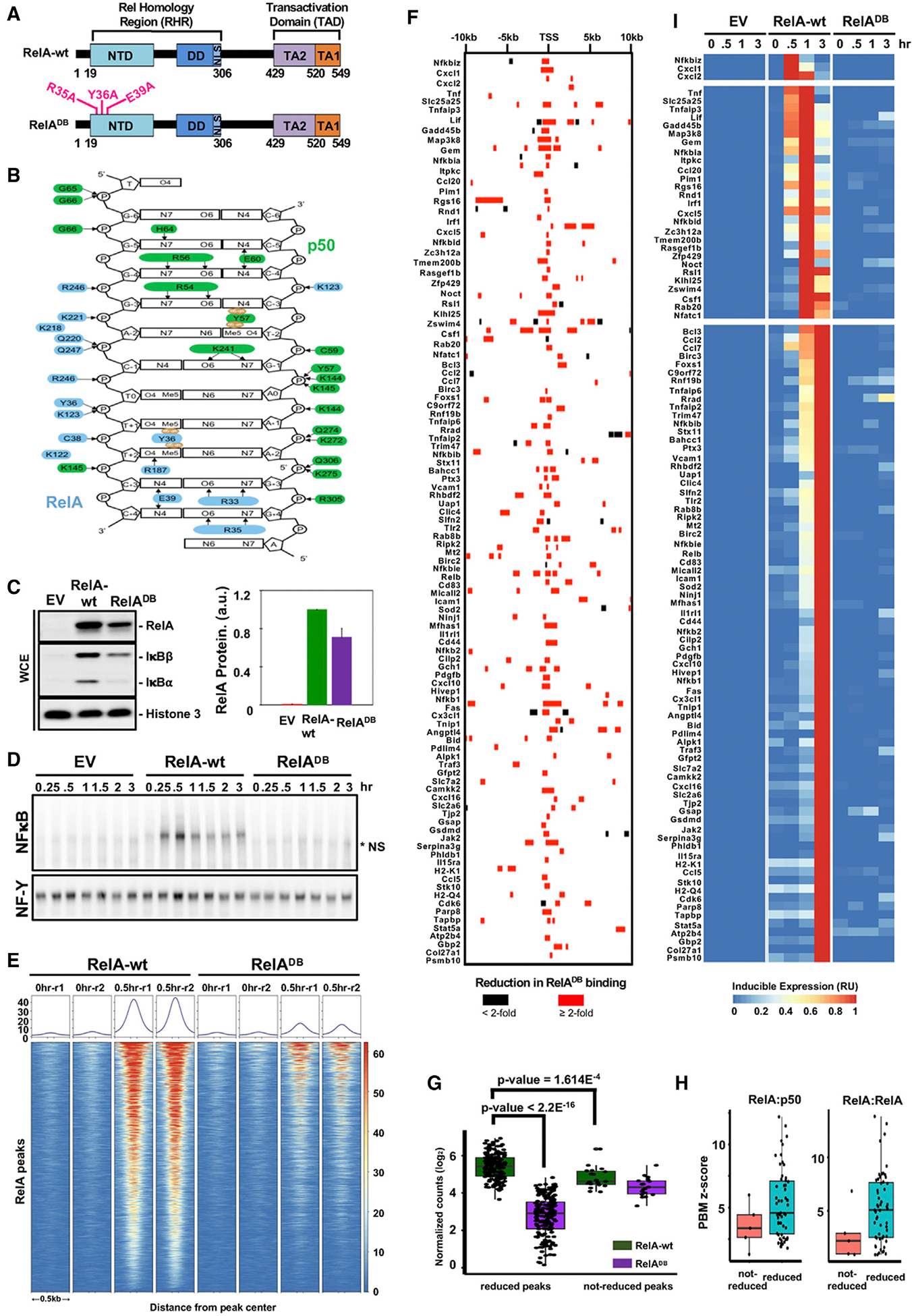
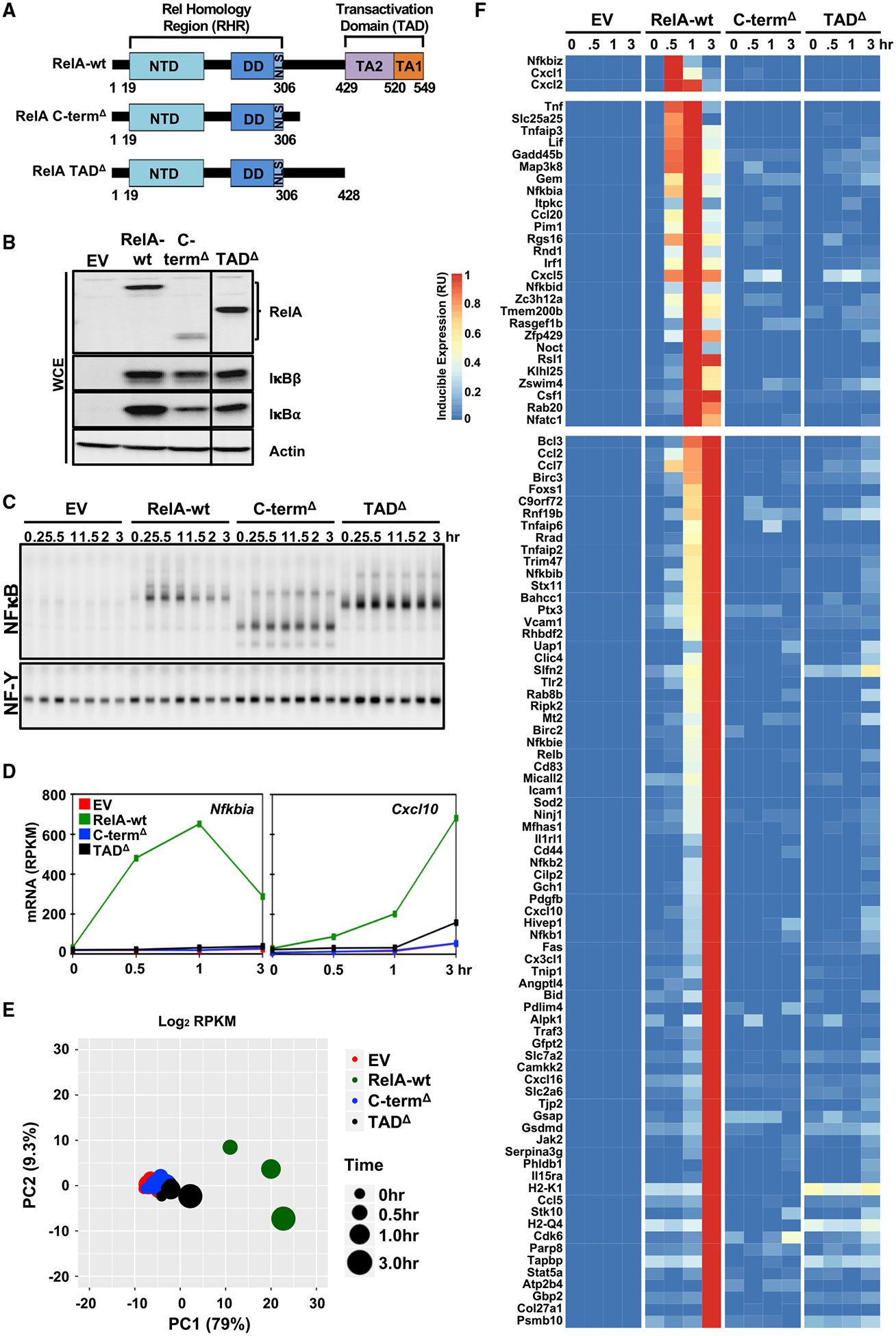
References
-
- Bae JS, Jang MK, Hong S, An WG, Choi YH, Kim HD, and Cheong J (2003). Phosphorylation of NF-κ B by calmodulin-dependent kinase IV activates anti-apoptotic gene expression. Biochem. Biophys. Res. Commun 305, 1094–1098. - PubMed
-
- Baeuerle PA, and Baltimore D (1989). A 65-kD subunit of active NF-κB is required for inhibition of NF-κB by I κB. Genes Dev 3, 1689–1698. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
