

The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems
- PMID: 32102216
- PMCID: PMC7074908
- DOI: 10.3390/microorganisms8020308
The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems
Abstract
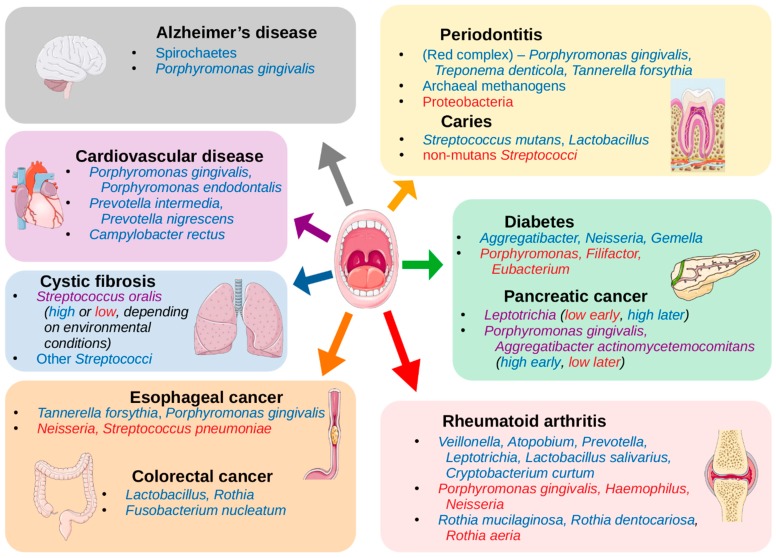
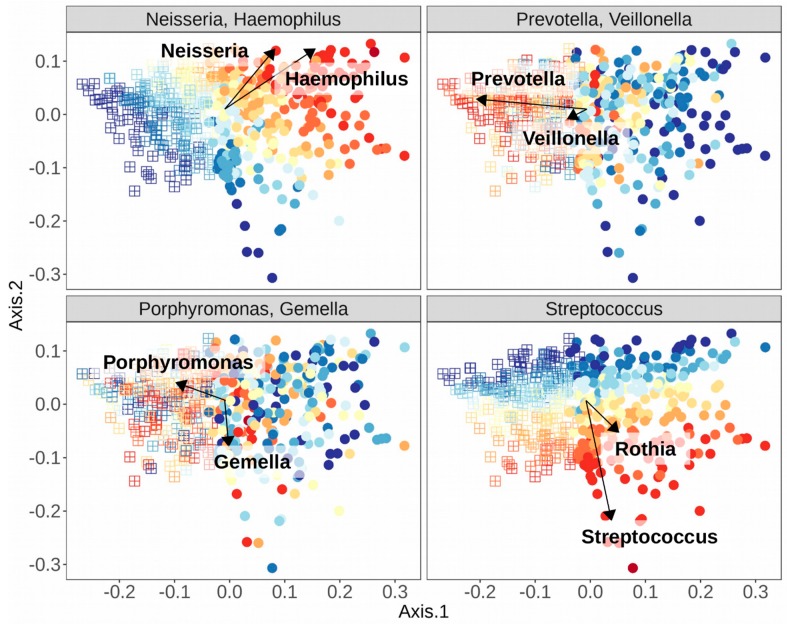
Abstract: The human oral cavity is home to an abundant and diverse microbial community (i.e., the oral microbiome), whose composition and roles in health and disease have been the focus of intense research in recent years. Thanks to developments in sequencing-based approaches, such as 16S ribosomal RNA metabarcoding, whole metagenome shotgun sequencing, or meta-transcriptomics, we now can efficiently explore the diversity and roles of oral microbes, even if unculturable. Recent sequencing-based studies have charted oral ecosystems and how they change due to lifestyle or disease conditions. As studies progress, there is increasing evidence of an important role of the oral microbiome in diverse health conditions, which are not limited to diseases of the oral cavity. This, in turn, opens new avenues for microbiome-based diagnostics and therapeutics that benefit from the easy accessibility of the oral cavity for microbiome monitoring and manipulation. Yet, many challenges remain ahead. In this review, we survey the main sequencing-based methodologies that are currently used to explore the oral microbiome and highlight major findings enabled by these approaches. Finally, we discuss future prospects in the field.
Keywords: Next generation sequencing; Oral microbiome; microbiome perturbations; oral diseases; stomatotypes; systemic diseases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
