

Discovery of Small-Molecule Activators for Glucose-6-Phosphate Dehydrogenase (G6PD) Using Machine Learning Approaches
- PMID: 32102234
- PMCID: PMC7073180
- DOI: 10.3390/ijms21041523
Discovery of Small-Molecule Activators for Glucose-6-Phosphate Dehydrogenase (G6PD) Using Machine Learning Approaches
Abstract
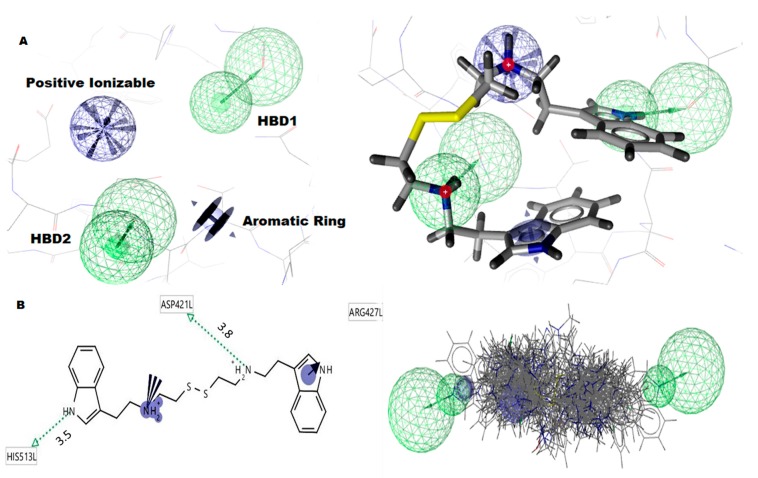
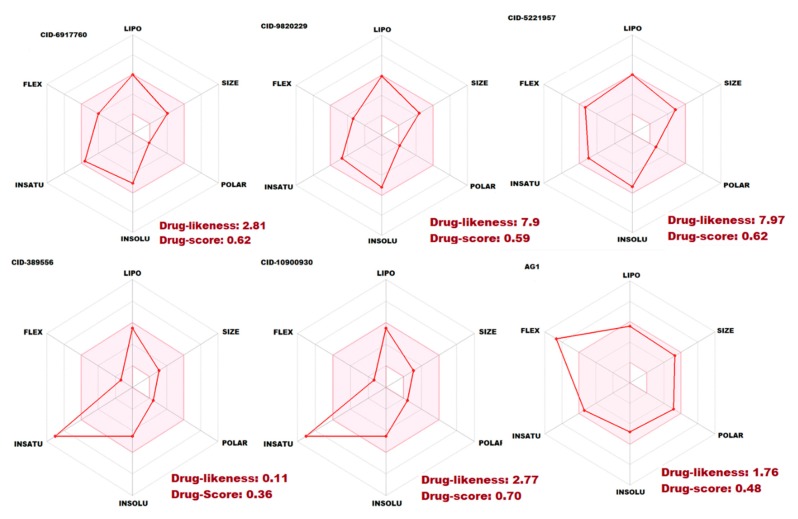
Glucose-6-Phosphate Dehydrogenase (G6PD) is a ubiquitous cytoplasmic enzyme converting glucose-6-phosphate into 6-phosphogluconate in the pentose phosphate pathway (PPP). The G6PD deficiency renders the inability to regenerate glutathione due to lack of Nicotine Adenosine Dinucleotide Phosphate (NADPH) and produces stress conditions that can cause oxidative injury to photoreceptors, retinal cells, and blood barrier function. In this study, we constructed pharmacophore-based models based on the complex of G6PD with compound AG1 (G6PD activator) followed by virtual screening. Fifty-three hit molecules were mapped with core pharmacophore features. We performed molecular descriptor calculation, clustering, and principal component analysis (PCA) to pharmacophore hit molecules and further applied statistical machine learning methods. Optimal performance of pharmacophore modeling and machine learning approaches classified the 53 hits as drug-like (18) and nondrug-like (35) compounds. The drug-like compounds further evaluated our established cheminformatics pipeline (molecular docking and in silico ADMET (absorption, distribution, metabolism, excretion and toxicity) analysis). Finally, five lead molecules with different scaffolds were selected by binding energies and in silico ADMET properties. This study proposes that the combination of machine learning methods with traditional structure-based virtual screening can effectively strengthen the ability to find potential G6PD activators used for G6PD deficiency diseases. Moreover, these compounds can be considered as safe agents for further validation studies at the cell level, animal model, and even clinic setting.
Keywords: ADMET; G6PD; docking; machine learning; pharmacophore modeling.
Conflict of interest statement
The authors have no competing interests.
Figures






References
-
- Carvalho C.G., Castro S.M., Santin A.P., Zaleski C., Carvalho F.G., Giugliani R. Glucose-6-phosphate-dehydrogenase deficiency and its correlation with other risk factors in jaundiced newborns in southern brazil. Asian Pac. J. Trop. Biomed. 2011;1:110–113. doi: 10.1016/S2221-1691(11)60006-3. - DOI - PMC - PubMed
-
- Naylor C.E., Rowland P., Basak A.K., Gover S., Mason P.J., Bautista J.M., Vulliamy T.J., Luzzatto L., Adams M.J. Glucose 6-phosphate dehydrogenase mutations causing enzyme deficiency in a model of the tertiary structure of the human enzyme. Blood. 1996;87:2974–2982. doi: 10.1182/blood.V87.7.2974.bloodjournal8772974. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
