

Identification of Alzheimer's disease-associated rare coding variants in the ECE2 gene
- PMID: 32102983
- PMCID: PMC7101146
- DOI: 10.1172/jci.insight.135119
Identification of Alzheimer's disease-associated rare coding variants in the ECE2 gene
Abstract
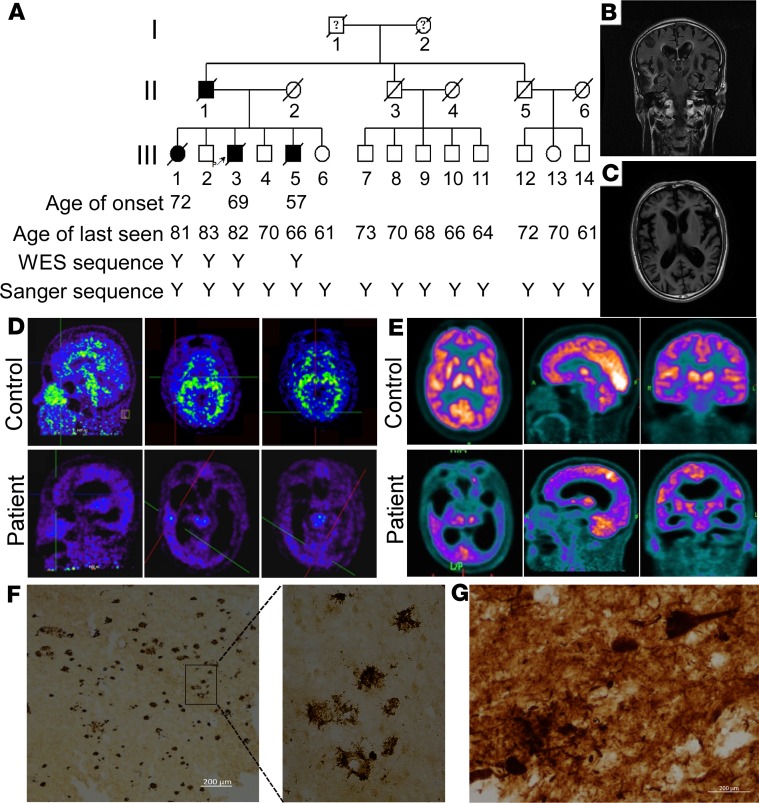
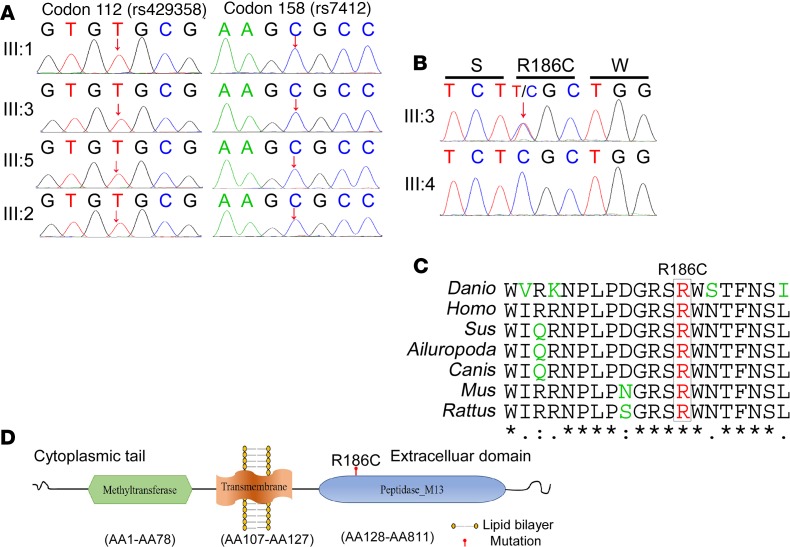
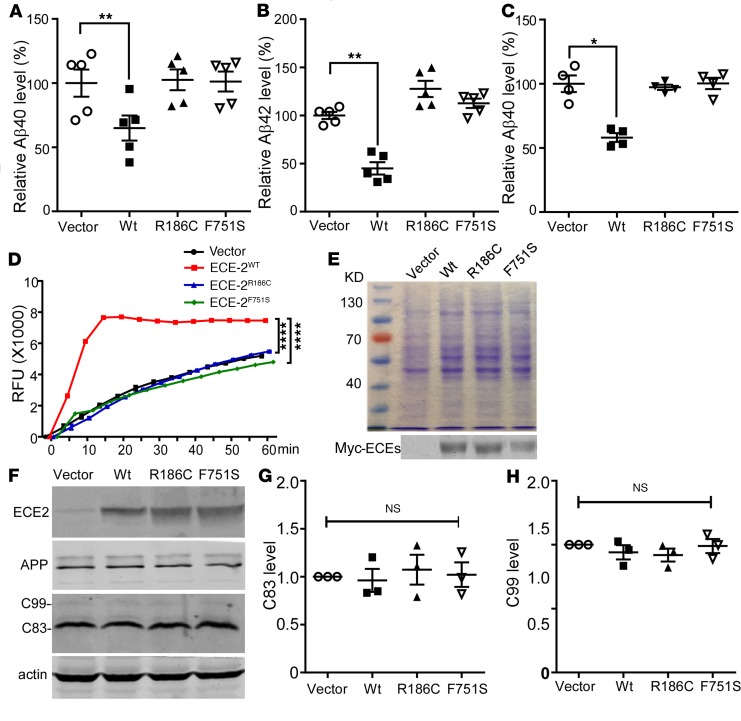
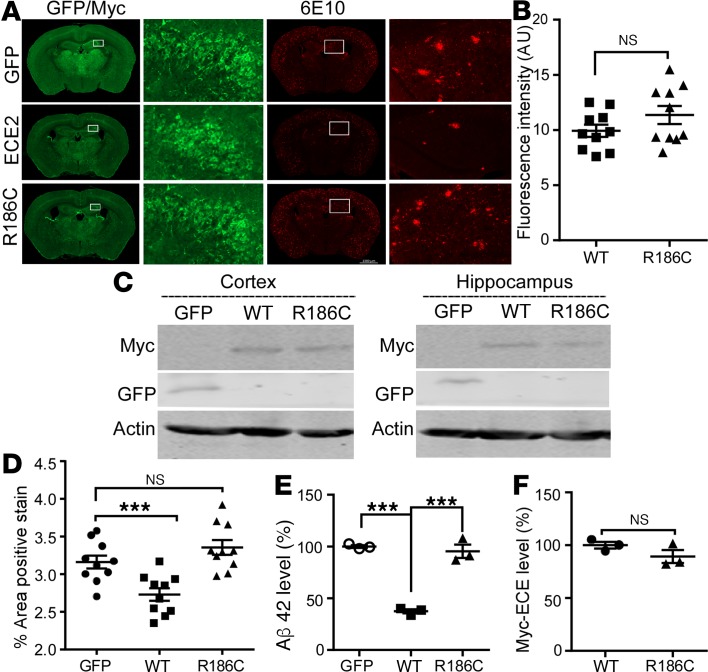
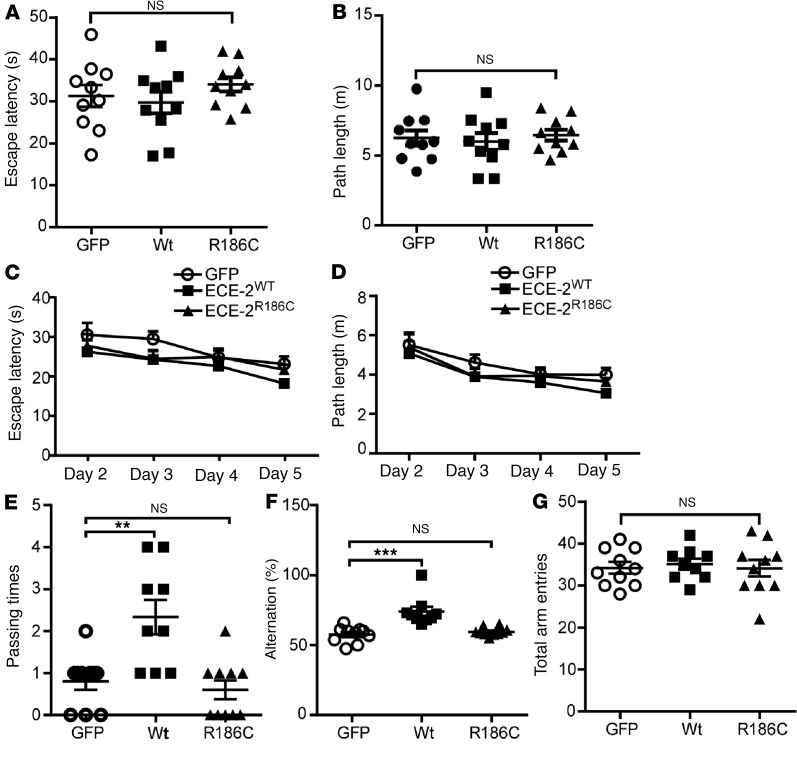
Accumulation of amyloid β protein (Aβ) due to increased generation and/or impaired degradation plays an important role in Alzheimer's disease (AD) pathogenesis. In this report, we describe the identification of rare coding mutations in the endothelin-converting enzyme 2 (ECE2) gene in 1 late-onset AD family, and additional case-control cohort analysis indicates ECE2 variants associated with the risk of developing AD. The 2 mutations (R186C and F751S) located in the peptidase domain in the ECE2 protein were found to severely impair the enzymatic activity of ECE2 in Aβ degradation. We further evaluated the effect of the R186C mutation in mutant APP-knockin mice. Overexpression of wild-type ECE2 in the hippocampus reduced amyloid load and plaque formation, and improved learning and memory deficits in the AD model mice. However, the effect was abolished by the R186C mutation in ECE2. Taken together, the results demonstrated that ECE2 peptidase mutations contribute to AD pathogenesis by impairing Aβ degradation, and overexpression of ECE2 alleviates AD phenotypes. This study indicates that ECE2 is a risk gene for AD development and pharmacological activation of ECE2 could be a promising strategy for AD treatment.
Keywords: Alzheimer’s disease; Genetic variation; Neuroscience.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
