

Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG
- PMID: 32103184
- PMCID: PMC7275909
- DOI: 10.1038/s41436-020-0767-8
Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG
Abstract
Purpose: We studied galactose supplementation in SLC35A2-congenital disorder of glycosylation (SLC35A2-CDG), caused by monoallelic pathogenic variants in SLC35A2 (Xp11.23), encoding the endoplasmic reticulum (ER) and Golgi UDP-galactose transporter. Patients present with epileptic encephalopathy, developmental disability, growth deficiency, and dysmorphism.
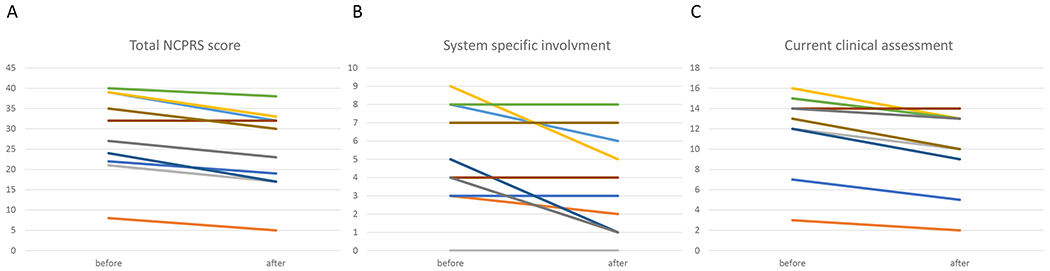
Methods: Ten patients with SLC35A2-CDG were supplemented with oral D-galactose for 18 weeks in escalating doses up to 1.5 g/kg/day. Outcome was assessed using the Nijmegen Pediatric CDG Rating Scale (NPCRS, ten patients) and by glycomics (eight patients).
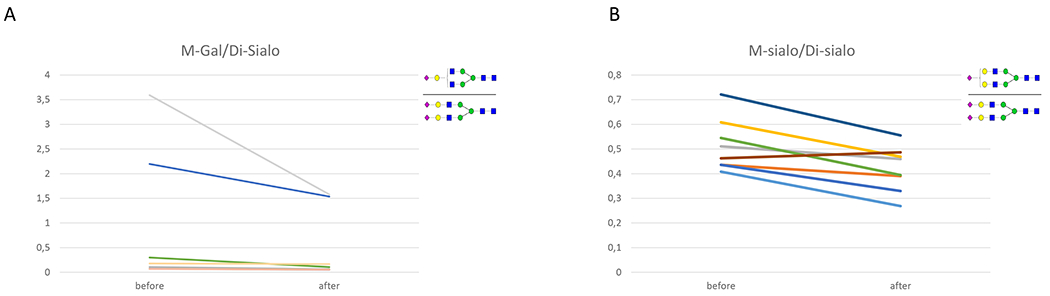
Results: SLC35A2-CDG patients demonstrated improvements in overall Nijmegen Pediatric CDG Rating Scale (NPCRS) score (P = 0.008), the current clinical assessment (P = 0.007), and the system specific involvement (P = 0.042) domains. Improvements were primarily in growth and development with five patients resuming developmental progress, which included postural control, response to stimuli, and chewing and swallowing amelioration. Additionally, there were improvements in gastrointestinal symptoms and epilepsy. One patient in our study did not show any clinical improvement. Galactose supplementation improved patients' glycosylation with decreased ratios of incompletely formed to fully formed glycans (M-gal/disialo, P = 0.012 and monosialo/disialo, P = 0.017) and increased levels of a fully galactosylated N-glycan (P = 0.05).
Conclusions: Oral D-galactose supplementation results in clinical and biochemical improvement in SLC35A2-CDG. Galactose supplementation may partially overcome the Golgi UDP-galactose deficiency and improves galactosylation. Oral galactose is well tolerated and shows promise as dietary therapy.
Keywords: Nijmegen Pediatric CDG Rating Scale (NPCRS); SLC35A2-CDG; galactose; glycan; glycosylation.
Conflict of interest statement
Conflict of interests
There are no conflicts of interests for any of the authors.
Figures
References
-
- Kodera H, Nakamura K, Osaka H, et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum Mutat. 2013;34:1708–1714. - PubMed
-
- Dörre K, Olczak M, Wada Y, et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015;38:931–940. - PubMed
-
- Kimizu T, Takahashi Y, Oboshi T, et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev. 2017;39:256–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
