

Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2
- PMID: 32104911
- PMCID: PMC7228310
- DOI: 10.1002/jmv.25731
Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2
Abstract
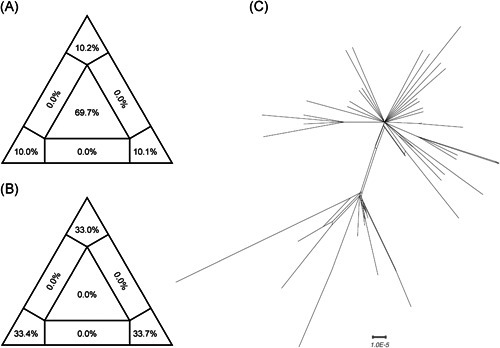
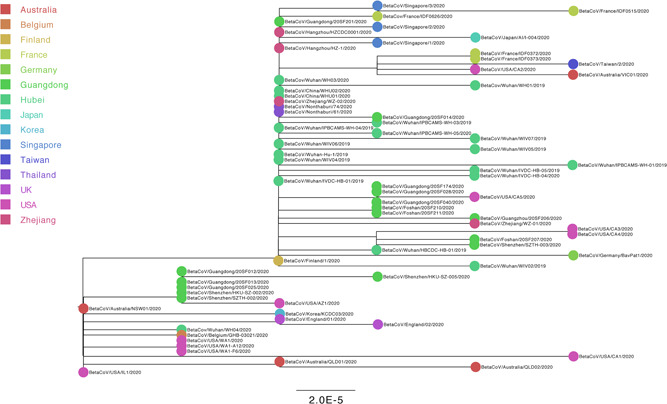
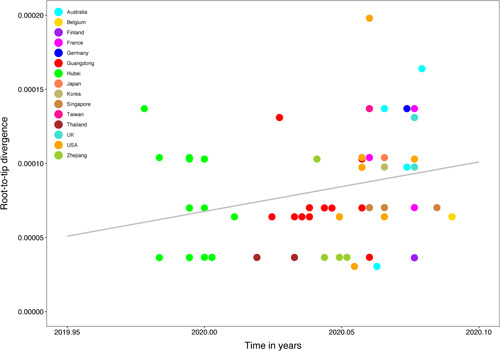
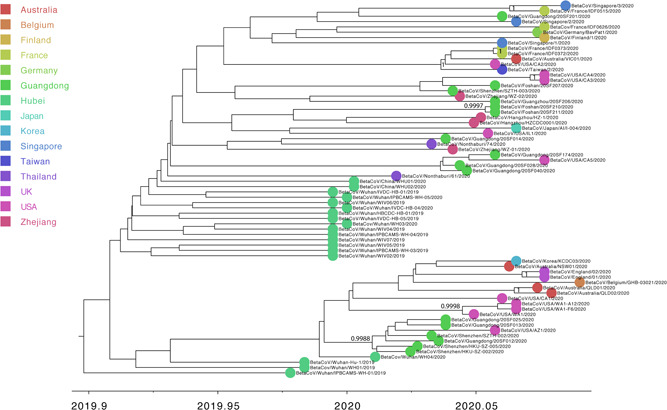
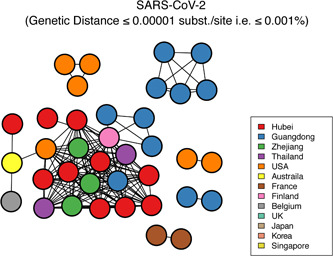
To investigate the evolutionary history of the recent outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in China, a total of 70 genomes of virus strains from China and elsewhere with sampling dates between 24 December 2019 and 3 February 2020 were analyzed. To explore the potential intermediate animal host of the SARS-CoV-2 virus, we reanalyzed virome data sets from pangolins and representative SARS-related coronaviruses isolates from bats, with particular attention paid to the spike glycoprotein gene. We performed phylogenetic, split network, transmission network, likelihood-mapping, and comparative analyses of the genomes. Based on Bayesian time-scaled phylogenetic analysis using the tip-dating method, we estimated the time to the most recent common ancestor and evolutionary rate of SARS-CoV-2, which ranged from 22 to 24 November 2019 and 1.19 to 1.31 × 10-3 substitutions per site per year, respectively. Our results also revealed that the BetaCoV/bat/Yunnan/RaTG13/2013 virus was more similar to the SARS-CoV-2 virus than the coronavirus obtained from the two pangolin samples (SRR10168377 and SRR10168378). We also identified a unique peptide (PRRA) insertion in the human SARS-CoV-2 virus, which may be involved in the proteolytic cleavage of the spike protein by cellular proteases, and thus could impact host range and transmissibility. Interestingly, the coronavirus carried by pangolins did not have the RRAR motif. Therefore, we concluded that the human SARS-CoV-2 virus, which is responsible for the recent outbreak of COVID-19, did not come directly from pangolins.
Keywords: COVID-19; SARS-CoV-2; TMRCA; cross-species transmission; evolutionary rate; potential intermediate animal host.
© 2020 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare that there are no conflict of interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
