

Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells
- PMID: 32106243
- PMCID: PMC7046262
- DOI: 10.1371/journal.pone.0229104
Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells
Abstract
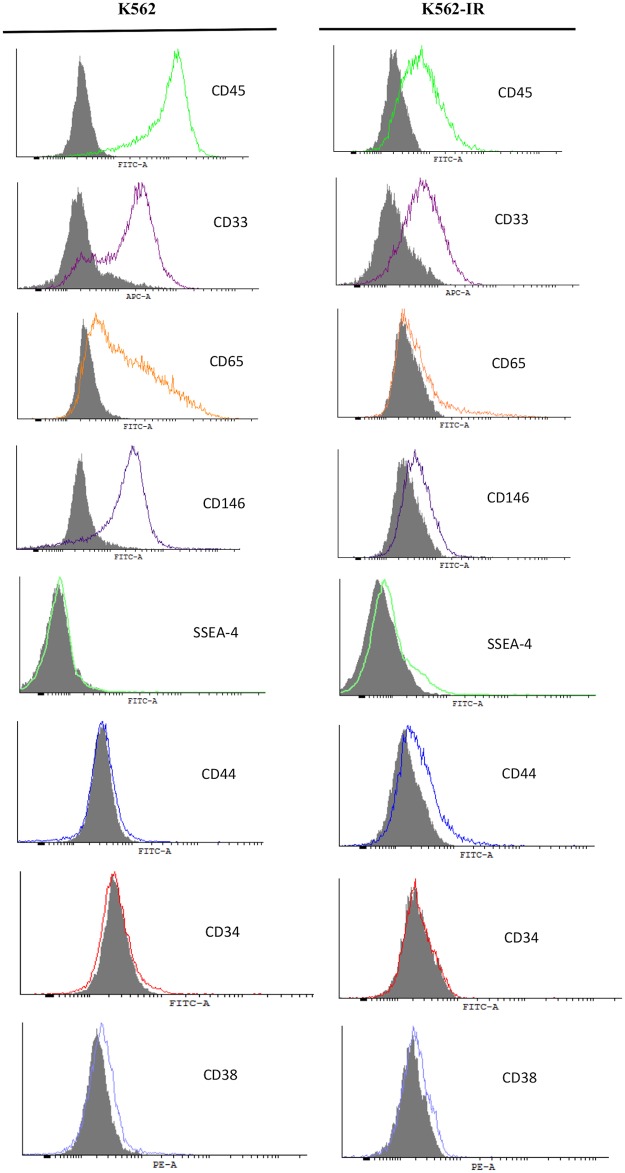
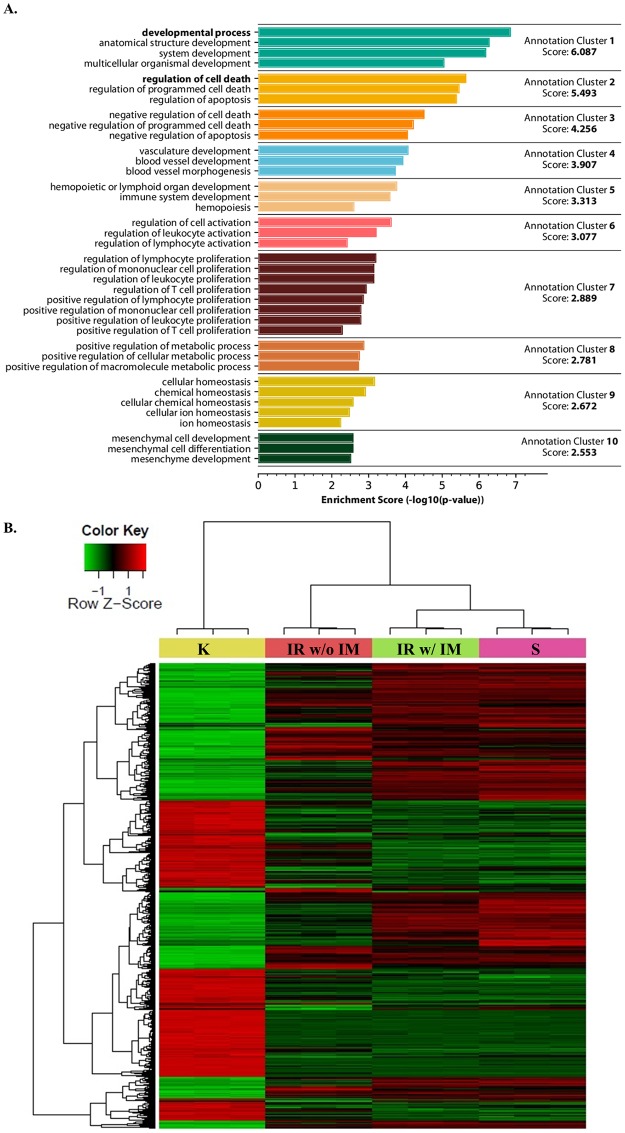
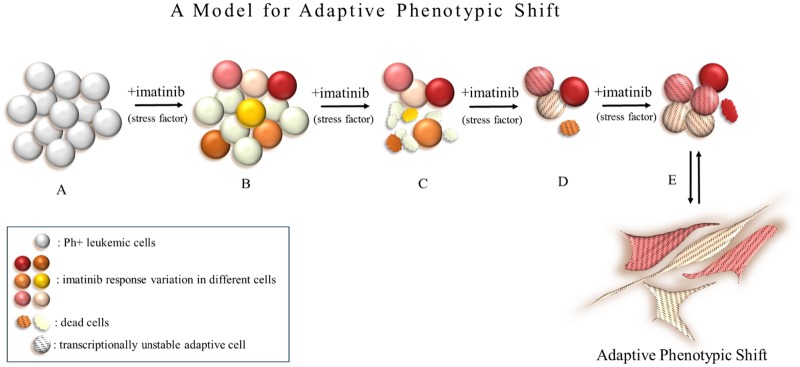
Tyrosine kinase inhibitor (TKI) resistance is a major problem in chronic myeloid leukemia (CML). We generated a TKI-resistant K562 sub-population, K562-IR, under selective imatinib-mesylate pressure. K562-IR cells are CD34-/CD38-, BCR-Abl-independent, proliferate slowly, highly adherent and form intact tumor spheroids. Loss of CD45 and other hematopoietic markers reveal these cells have diverged from their hematopoietic origin. CD34 negativity, high expression of E-cadherin and CD44; decreased levels of CD45 and β-catenin do not fully confer with the leukemic stem cell (LSC) phenotype. Expression analyses reveal that K562-IR cells differentially express tissue/organ development and differentiation genes. Our data suggest that the observed phenotypic shift is an adaptive process rendering cells under TKI stress to become oncogene independent. Cells develop transcriptional instability in search for a gene expression framework suitable for new environmental stresses, resulting in an adaptive phenotypic shift in which some cells partially display LSC-like properties. With leukemic/cancer stem cell targeted therapies underway, the difference between treating an entity and a spectrum of dynamic cellular states will have conclusive effects on the outcome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
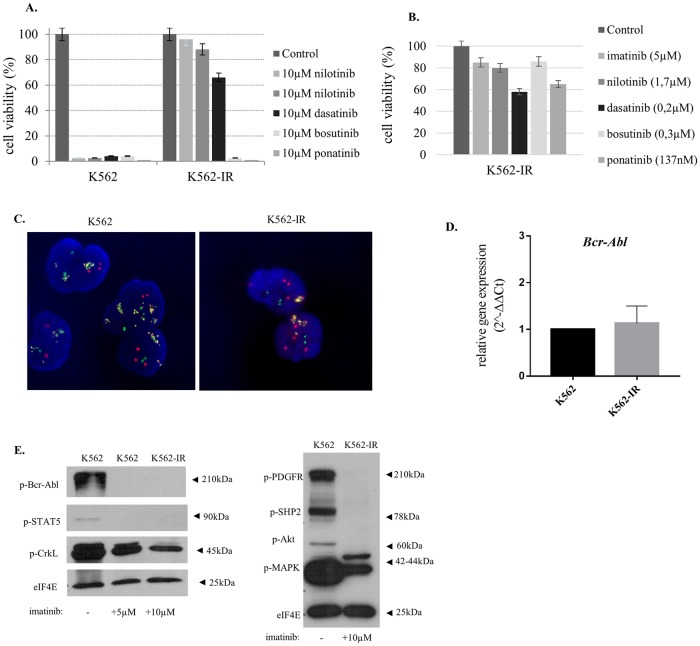
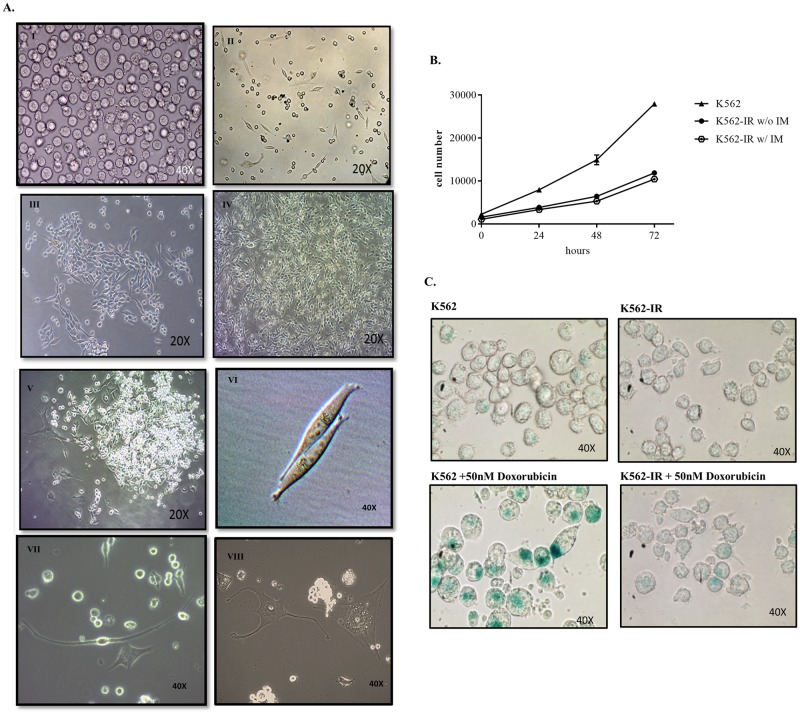
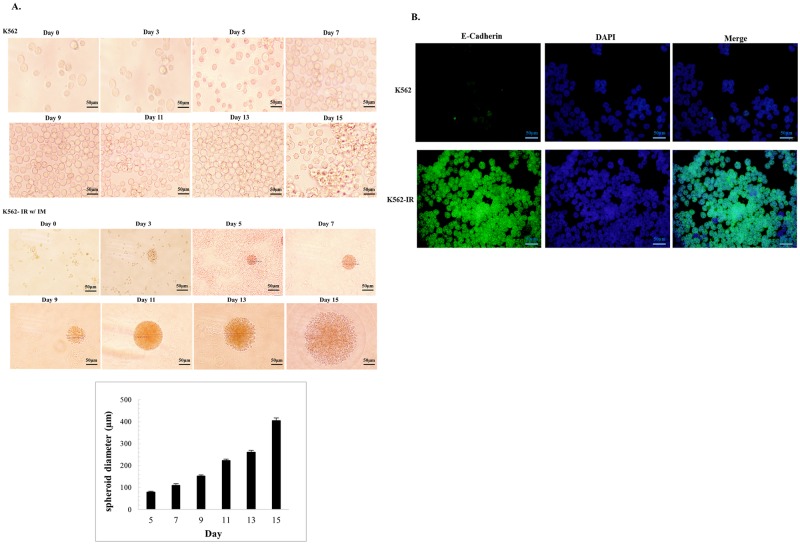
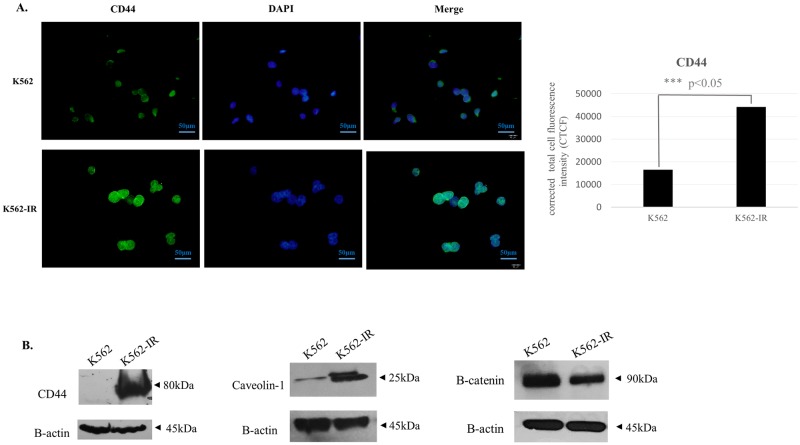
References
-
- Deininger MWN, Goldman JM, Melo JV, Dc W, Deininger MWN, Goldman JM, et al. The molecular biology of chronic myeloid leukemia Review article The molecular biology of chronic myeloid leukemia. 2000;96: 3343–3356. - PubMed
-
- Assouline S, Lipton JH. Monitoring response and resistance to treatment in chronic myeloid leukemia. Curr Oncol. 2011;18: e71–83. Available: http://www.ncbi.nlm.nih.gov/pubmed/21505592 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
