

Genetic Modifiers and Rare Mendelian Disease
- PMID: 32106447
- PMCID: PMC7140819
- DOI: 10.3390/genes11030239
Genetic Modifiers and Rare Mendelian Disease
Abstract
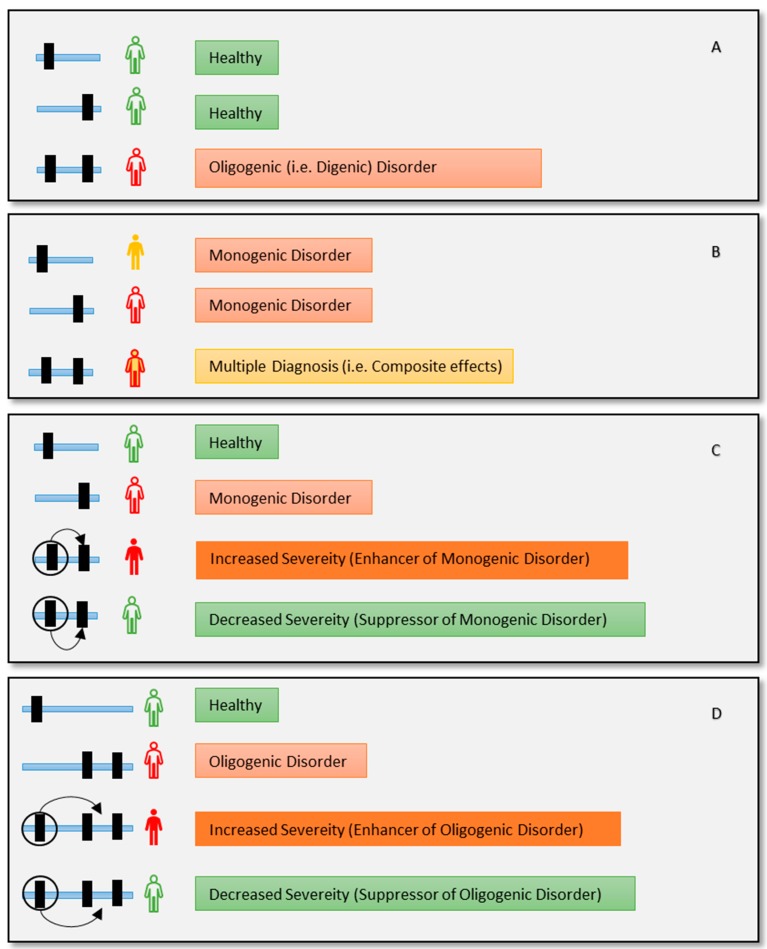
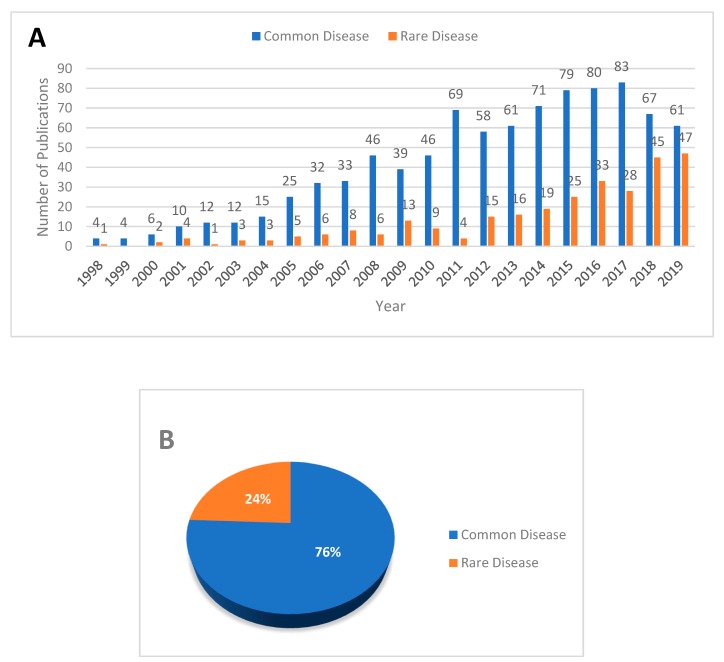
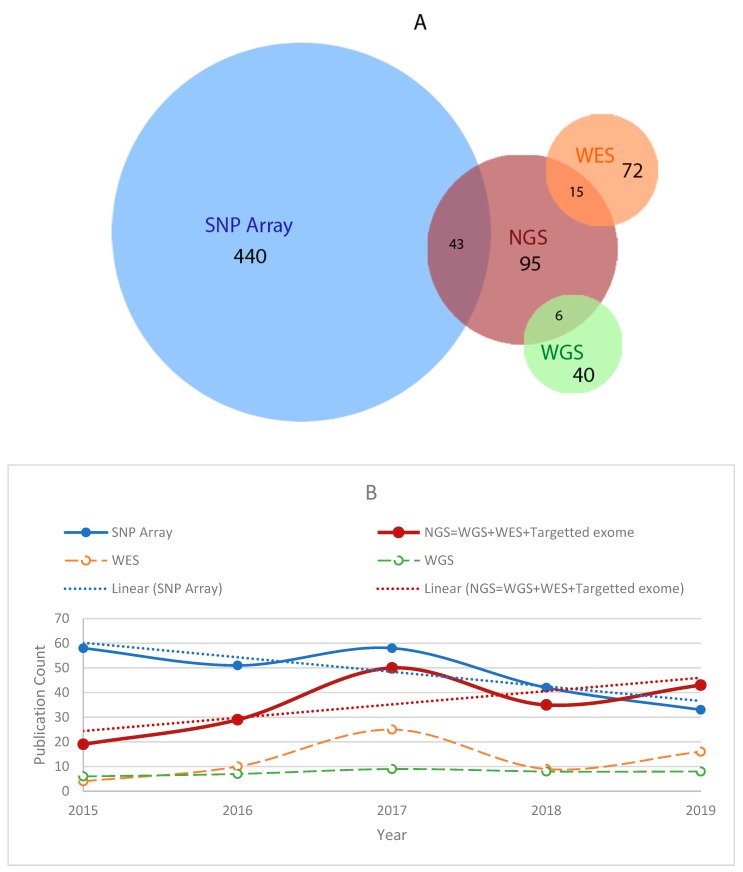
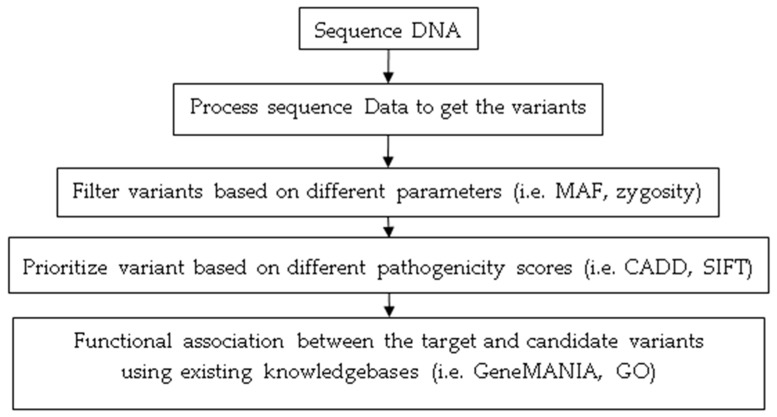
Despite advances in high-throughput sequencing that have revolutionized the discovery of gene defects in rare Mendelian diseases, there are still gaps in translating individual genome variation to observed phenotypic outcomes. While we continue to improve genomics approaches to identify primary disease-causing variants, it is evident that no genetic variant acts alone. In other words, some other variants in the genome (genetic modifiers) may alleviate (suppress) or exacerbate (enhance) the severity of the disease, resulting in the variability of phenotypic outcomes. Thus, to truly understand the disease, we need to consider how the disease-causing variants interact with the rest of the genome in an individual. Here, we review the current state-of-the-field in the identification of genetic modifiers in rare Mendelian diseases and discuss the potential for future approaches that could bridge the existing gap.
Keywords: GWAS; bioinformatics; expressivity; genetic interaction; genetic modifier; genome sequencing; mendelian disease; penetrance; phenotypic variability; rare disease.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Baltimore M. Online Mendelian Inheritance in Man, OMIM®: OMIM Entry Statistics. [(accessed on 26 July 2019)]; Available online: https://www.omim.org/statistics/entry.
-
- WHO|Genes and Human Diseases. [(accessed on 6 December 2019)]; Available online: https://www.who.int/genomics/public/geneticdiseases/en/index2.html.
-
- Boycott K.M., Rath A., Chong J.X., Hartley T., Alkuraya F.S., Baynam G., Brookes A.J., Brudno M., Carracedo A., den Dunnen J.T., et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017;100:695–705. doi: 10.1016/j.ajhg.2017.04.003. - DOI - PMC - PubMed
-
- De Vrueh R., Baekelandt E.R.F., De Haan J.M.H. Priority Medicines for Europe and the World 2013 Update. World Health Organization; Geneva, Switzerland: 2013. Rare Diseases (Background Paper 6.19)
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
