

Pediatric hemophagocytic lymphohistiocytosis
- PMID: 32107531
- PMCID: PMC8212354
- DOI: 10.1182/blood.2019000936
Pediatric hemophagocytic lymphohistiocytosis
Abstract
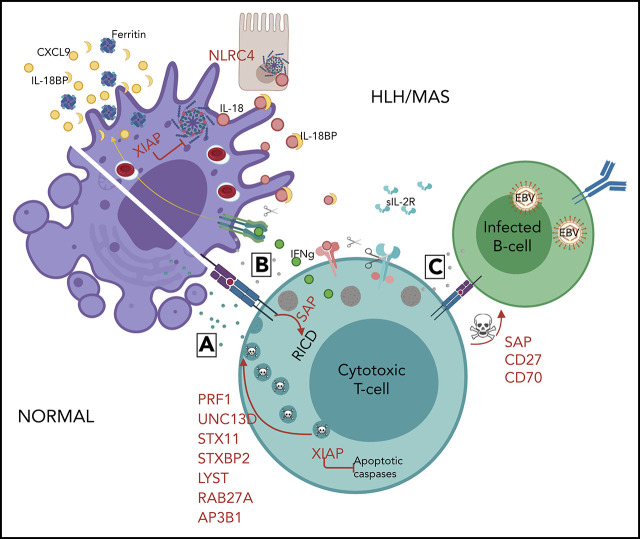
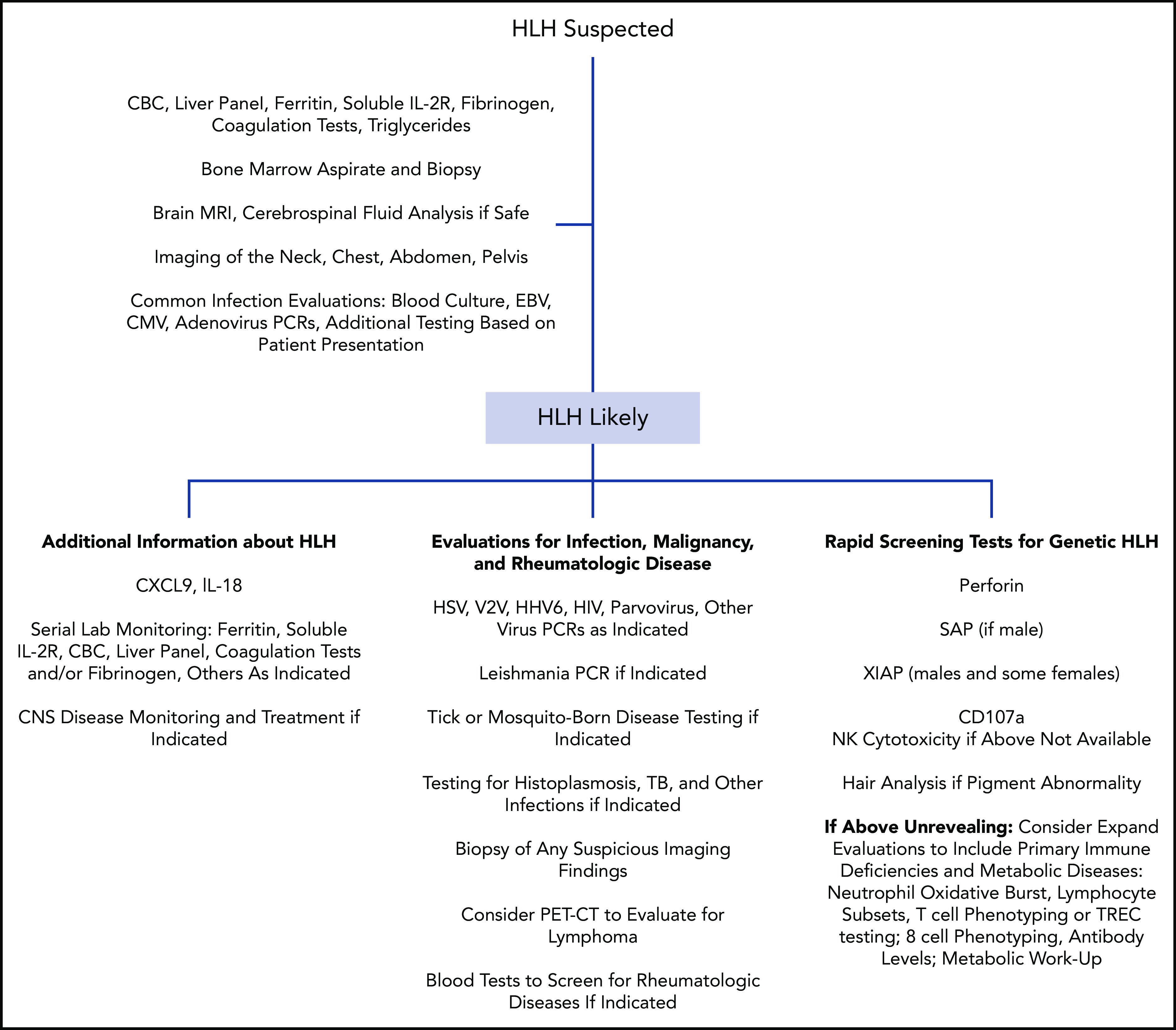
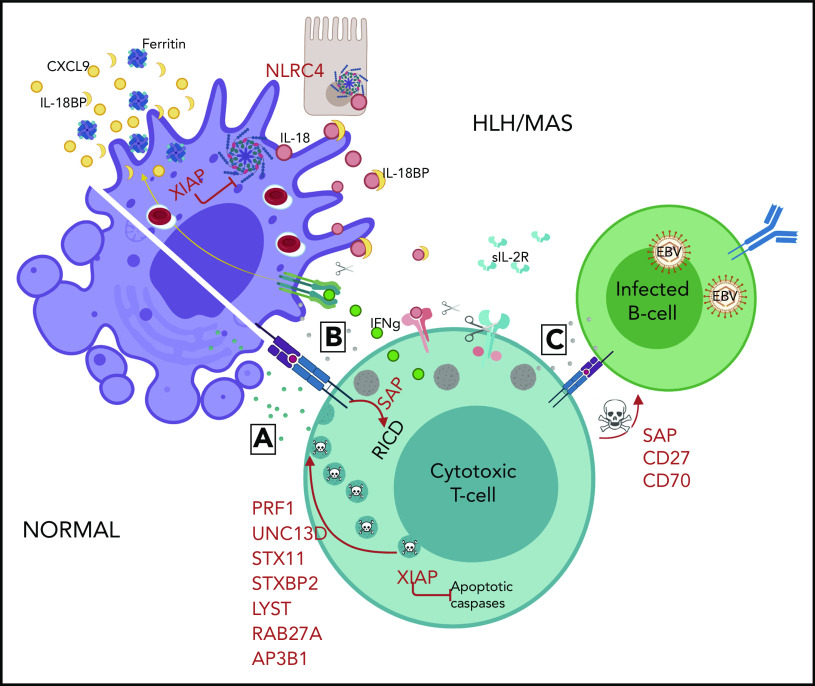
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome describing patients with severe systemic hyperinflammation. Characteristic features include unremitting fever, cytopenias, hepatosplenomegaly, and elevation of typical HLH biomarkers. Patients can develop hepatitis, coagulopathy, liver failure, central nervous system involvement, multiorgan failure, and other manifestations. The syndrome has a high mortality rate. More and more, it is recognized that while HLH can be appropriately used as a broad summary diagnosis, many pediatric patients actually suffer from an expanding spectrum of genetic diseases that can be complicated by the syndrome of HLH. Classic genetic diseases in which HLH is a typical and common manifestation include pathogenic changes in familial HLH genes (PRF1, UNC13D, STXBP2, and STX11), several granule/pigment abnormality genes (RAB27A, LYST, and AP3B1), X-linked lymphoproliferative disease genes (SH2D1A and XIAP), and others such as NLRC4, CDC42, and the Epstein-Barr virus susceptibility diseases. There are many other genetic diseases in which HLH is an infrequent complication of the disorder as opposed to a prominent manifestation of the disease caused directly by the genetic defect, including other primary immune deficiencies and inborn errors of metabolism. HLH can also occur in patients with underlying rheumatologic or autoinflammatory disorders and is usually designated macrophage activation syndrome in those settings. Additionally, HLH can develop in patients during infections or malignancies without a known (or as-yet-identified) genetic predisposition. This article will attempt to summarize current concepts in the pediatric HLH field as well as offer a practical diagnostic and treatment overview.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: S.W.C. has been a consultant for AB2Bio, Ltd. and received honoraria from Novartis, Inc. R.A.M. declares no competing financial interests.
Figures
References
-
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957-1959. - PubMed
-
- Sayos J, Wu C, Morra M, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395(6701):462-469. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
