

Pathobiology and treatment of lymphomatoid granulomatosis, a rare EBV-driven disorder
- PMID: 32107539
- PMCID: PMC7162687
- DOI: 10.1182/blood.2019000933
Pathobiology and treatment of lymphomatoid granulomatosis, a rare EBV-driven disorder
Abstract
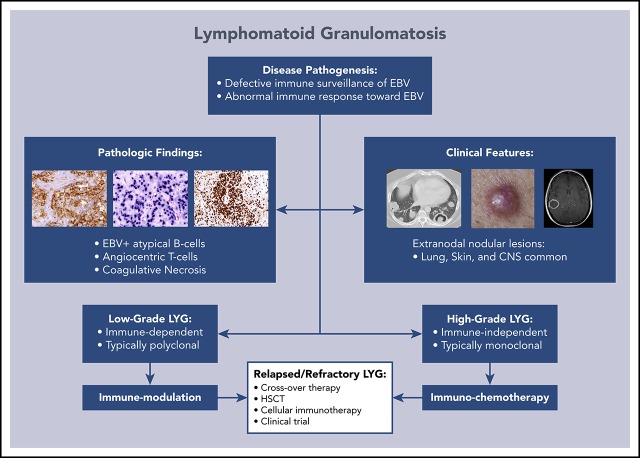
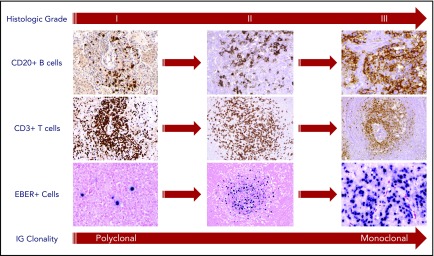
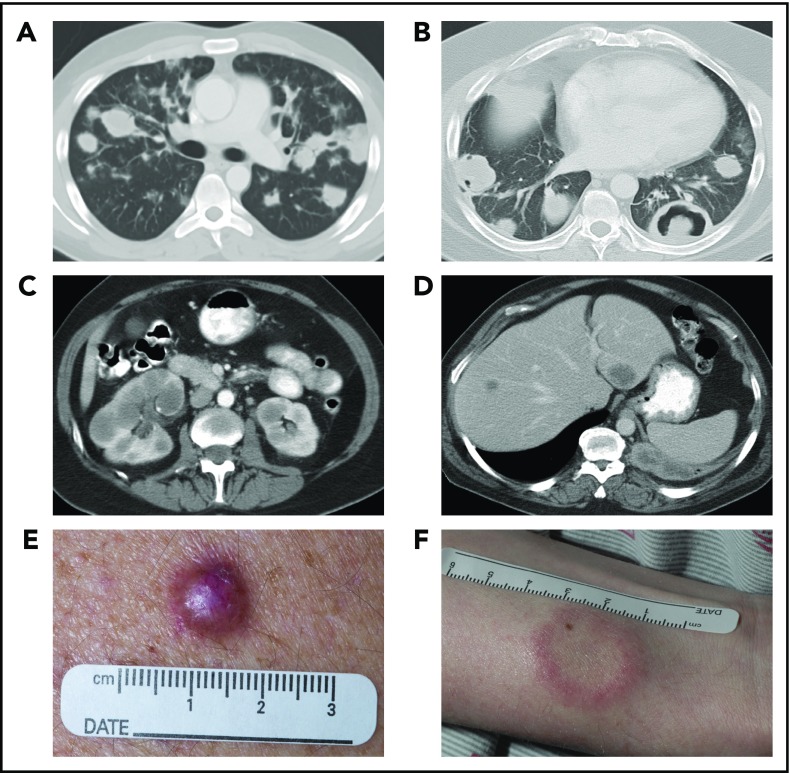
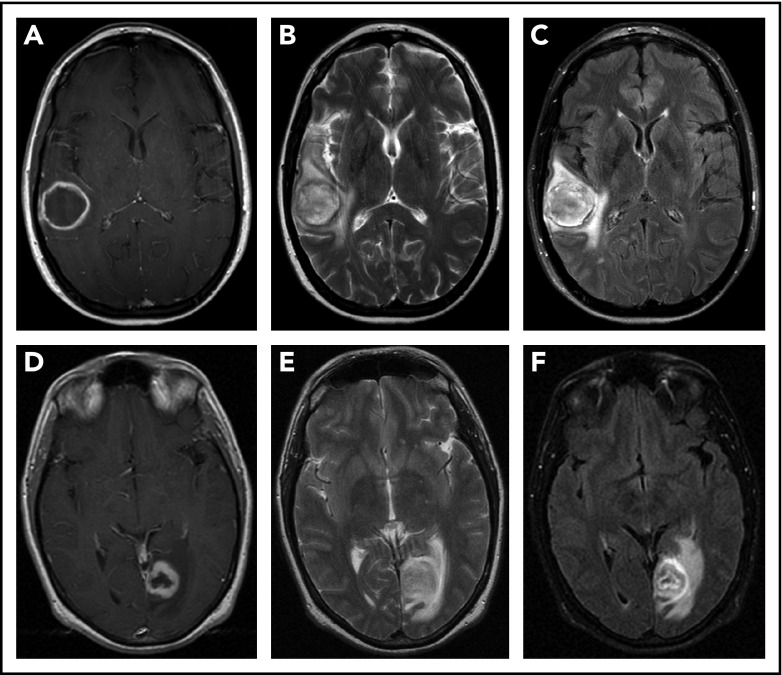
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)-driven B-cell lymphoproliferative disease (LPD). This disease is hypothesized to result from defective immune surveillance of EBV, with most patients showing evidence of immune dysfunction, despite no known primary immunodeficiency. Pathologically, LYG is graded by the number and density of EBV+ atypical B cells, and other characteristic findings include an angioinvasive/angiodestructive reactive T-cell infiltrate and various degrees of necrosis. Clinically, LYG universally involves the lungs with other common extranodal sites, including skin, central nervous system, liver, and kidneys. Nodal and/or bone marrow involvement is extremely rare and, if present, suggests an alternative diagnosis. Treatment selection is based on histologic grade and underlying pathobiology with low-grade disease hypothesized to be immune-dependent and typically polyclonal and high-grade disease to be immune-independent and typically oligoclonal or monoclonal. Methods of augmenting the immune response to EBV in low-grade LYG include treatment with interferon-α2b, whereas high-grade disease requires immunochemotherapy. Given the underlying defective immune surveillance of EBV, patients with high-grade disease may have a recurrence in the form of low-grade disease after immunochemotherapy, and those with low-grade disease may progress to high-grade disease after immune modulation, which can be effectively managed with crossover treatment. In patients with primary refractory disease or in those with multiple relapses, hematopoietic stem cell transplantation may be considered, but its efficacy is not well established. This review discusses the pathogenesis of LYG and highlights distinct histopathologic and clinical features that distinguish this disorder from other EBV+ B-cell LPDs and lymphomas. Treatment options, including immune modulation and combination immunochemotherapy, are discussed.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Liebow AA, Carrington CR, Friedman PJ. Lymphomatoid granulomatosis. Hum Pathol. 1972;3(4):457-558. - PubMed
-
- Nichols PW, Koss M, Levine AM, Lukes RJ. Lymphomatoid granulomatosis: a T-cell disorder? Am J Med. 1982;72(3):467-471. - PubMed
-
- Lipford EH Jr., Margolick JB, Longo DL, Fauci AS, Jaffe ES. Angiocentric immunoproliferative lesions: a clinicopathologic spectrum of post-thymic T-cell proliferations. Blood. 1988;72(5):1674-1681. - PubMed
-
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43(1):360-373. - PubMed
-
- Katzenstein AL, Peiper SC. Detection of Epstein-Barr virus genomes in lymphomatoid granulomatosis: analysis of 29 cases by the polymerase chain reaction technique. Mod Pathol. 1990;3(4):435-441. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
