

HDAC3 functions as a positive regulator in Notch signal transduction
- PMID: 32107550
- PMCID: PMC7144913
- DOI: 10.1093/nar/gkaa088
HDAC3 functions as a positive regulator in Notch signal transduction
Abstract
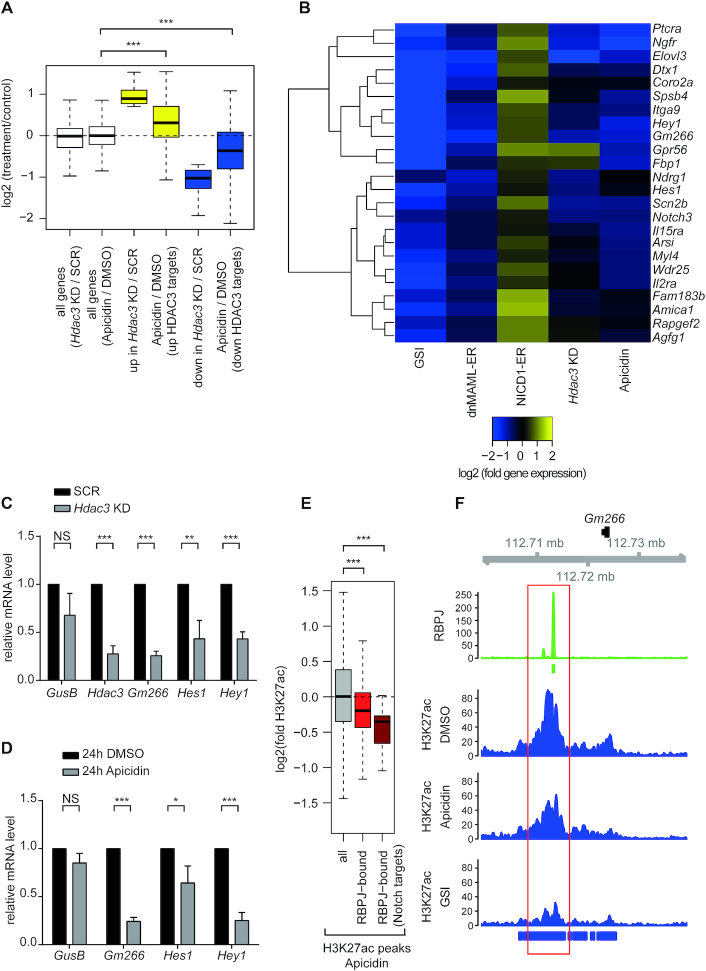
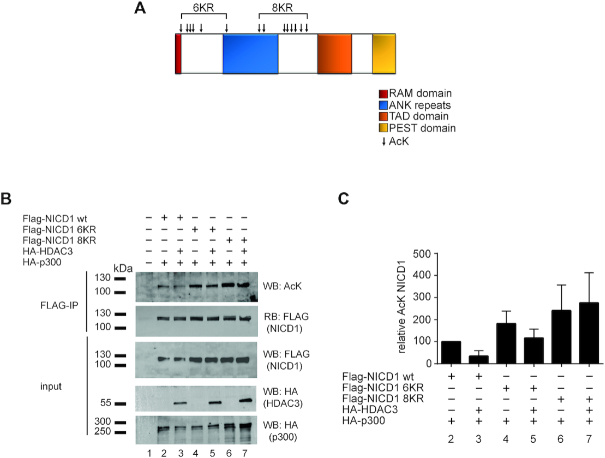
Aberrant Notch signaling plays a pivotal role in T-cell acute lymphoblastic leukemia (T-ALL) and chronic lymphocytic leukemia (CLL). Amplitude and duration of the Notch response is controlled by ubiquitin-dependent proteasomal degradation of the Notch1 intracellular domain (NICD1), a hallmark of the leukemogenic process. Here, we show that HDAC3 controls NICD1 acetylation levels directly affecting NICD1 protein stability. Either genetic loss-of-function of HDAC3 or nanomolar concentrations of HDAC inhibitor apicidin lead to downregulation of Notch target genes accompanied by a local reduction of histone acetylation. Importantly, an HDAC3-insensitive NICD1 mutant is more stable but biologically less active. Collectively, these data show a new HDAC3- and acetylation-dependent mechanism that may be exploited to treat Notch1-dependent leukemias.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
