

Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis
- PMID: 32108159
- PMCID: PMC7046743
- DOI: 10.1038/s41598-020-60015-4
Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis
Abstract
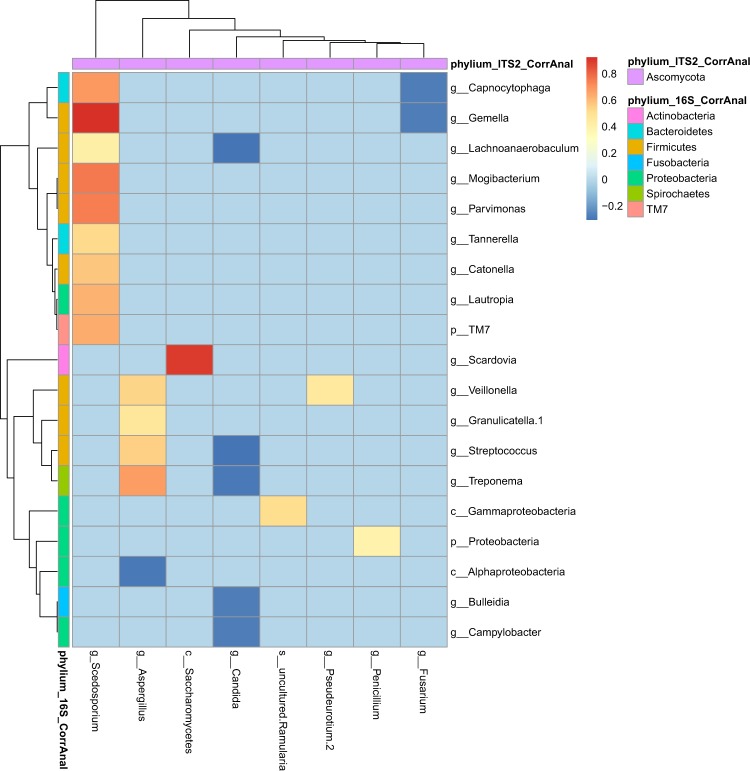
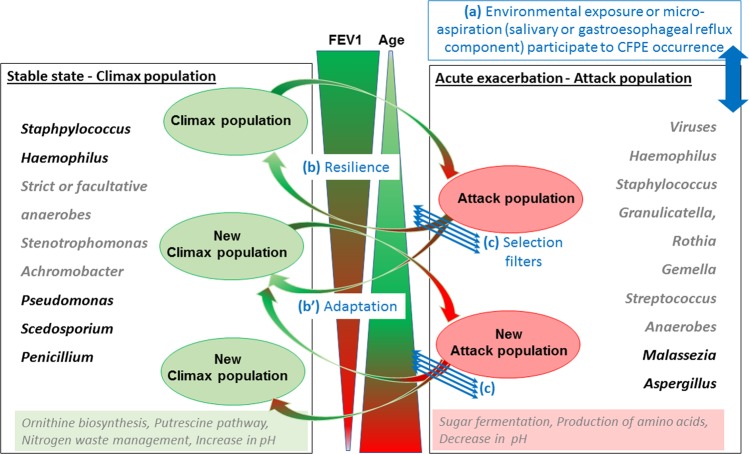
Lung infections play a critical role in cystic fibrosis (CF) pathogenesis. CF respiratory tract is now considered to be a polymicrobial niche and advances in high-throughput sequencing allowed to analyze its microbiota and mycobiota. However, no NGS studies until now have characterized both communities during CF pulmonary exacerbation (CFPE). Thirty-three sputa isolated from patients with and without CFPE were used for metagenomic high-throughput sequencing targeting 16S and ITS2 regions of bacterial and fungal rRNA. We built inter-kingdom network and adapted Phy-Lasso method to highlight correlations in compositional data. The decline in respiratory function was associated with a decrease in bacterial diversity. The inter-kingdom network revealed three main clusters organized around Aspergillus, Candida, and Scedosporium genera. Using Phy-Lasso method, we identified Aspergillus and Malassezia as relevantly associated with CFPE, and Scedosporium plus Pseudomonas with a decline in lung function. We corroborated in vitro the cross-domain interactions between Aspergillus and Streptococcus predicted by the correlation network. For the first time, we included documented mycobiome data into a version of the ecological Climax/Attack model that opens new lines of thoughts about the physiopathology of CF lung disease and future perspectives to improve its therapeutic management.
Conflict of interest statement
Pr L. Delhaes is member of the scientific advisory board of “Vaincre la Mucoviscidose”, and of the executive committee of ESGHAMI (ESCMID working group). She also did punctual consulting for pharmaceutical companies (Pfizer, Gilead, and MSD) and was invited to participate to scientific congresses as compensations. The other authors declare no potential conflict of interest.
Figures
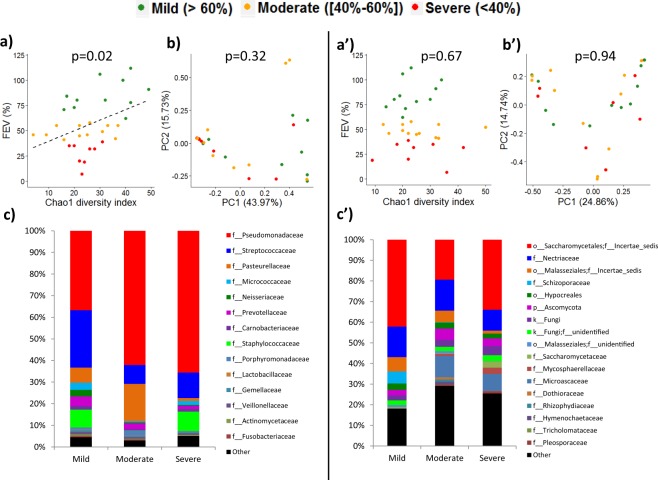
 ), moderate (
), moderate ( ) and severe (
) and severe ( ) alteration of the lung function measured by FEV1. Dataset from each sputum sample contained an average of 7,909 bacterial reads (ranging from 5,104 to 14,749), and an average of 9,811 fungal reads (ranging from 2,933 to 15,599). Alpha diversity indexes of the bacterial microbiome (a) but not of mycobiome (a′) were positively correlated with FEV1 values. When patients were divided into 3 groups according to FEV1 values,, bi-dimensional PCoA representations (b,b′) based on Bray-Curtis similarity matrix did not show clustering between groups. However, we observed a high proportion (62% and 66%) of Pseudomonadaceae (mainly composed of Pseudomonas species) among patients exhibiting a moderate and severe disease, while it represents only 32% of the bacterial composition among patients with a mild ventilatory deficit (c). The fungal composition exhibited also some shifts, especially a decrease in OTUs belonging to Malasseziales in samples from patients with severe lung decline (c′). Taxonomy composition were represented at family level of bacterial (c) and fungal (c′) microbiotas; fungal reads that were not identified as family levels are grouped at order or phylum levels.
) alteration of the lung function measured by FEV1. Dataset from each sputum sample contained an average of 7,909 bacterial reads (ranging from 5,104 to 14,749), and an average of 9,811 fungal reads (ranging from 2,933 to 15,599). Alpha diversity indexes of the bacterial microbiome (a) but not of mycobiome (a′) were positively correlated with FEV1 values. When patients were divided into 3 groups according to FEV1 values,, bi-dimensional PCoA representations (b,b′) based on Bray-Curtis similarity matrix did not show clustering between groups. However, we observed a high proportion (62% and 66%) of Pseudomonadaceae (mainly composed of Pseudomonas species) among patients exhibiting a moderate and severe disease, while it represents only 32% of the bacterial composition among patients with a mild ventilatory deficit (c). The fungal composition exhibited also some shifts, especially a decrease in OTUs belonging to Malasseziales in samples from patients with severe lung decline (c′). Taxonomy composition were represented at family level of bacterial (c) and fungal (c′) microbiotas; fungal reads that were not identified as family levels are grouped at order or phylum levels.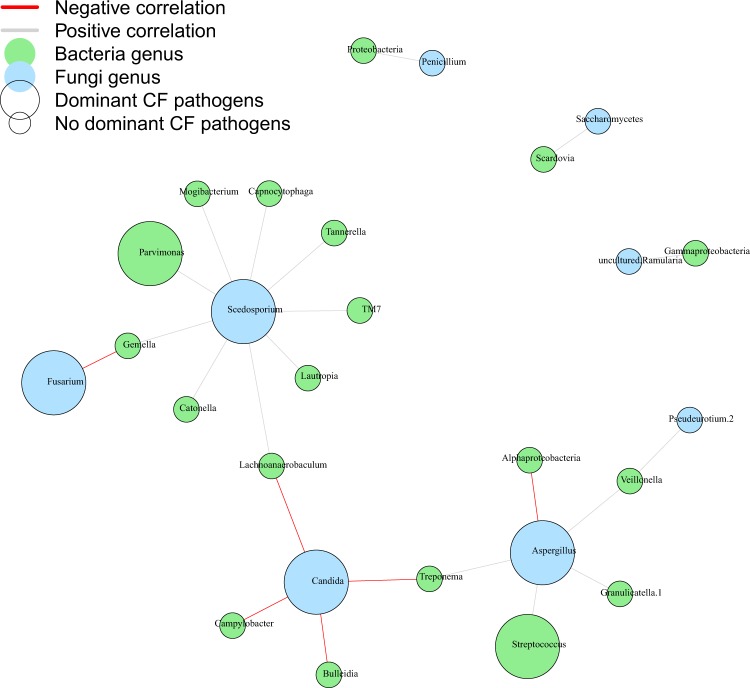

 ) expressed in CFU/mL is significantly enhanced by S. mitis at day 4 (p < 0.05) and day 5 (p < 0.01). Growth of A. fumigatus plus S. mitis (
) expressed in CFU/mL is significantly enhanced by S. mitis at day 4 (p < 0.05) and day 5 (p < 0.01). Growth of A. fumigatus plus S. mitis ( ) is significantly higher than growth of A. fumigatus plus S. oralis (
) is significantly higher than growth of A. fumigatus plus S. oralis ( ) at day 5 (p < 0.05). Growth of A. fumigatus was not significantly enhanced by S. oralis excepted at day 2 (p < 0.05).
) at day 5 (p < 0.05). Growth of A. fumigatus was not significantly enhanced by S. oralis excepted at day 2 (p < 0.05).
References
-
- Rush, S. T., Lee, C. H., Mio, W. & Kim, P. T. The Phylogenetic LASSO and the Microbiome. ArXiv160708877 Q-Bio Stat (2016).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
