

Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes
- PMID: 32108862
- PMCID: PMC7112083
- DOI: 10.1093/bioinformatics/btaa145
Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes
Abstract
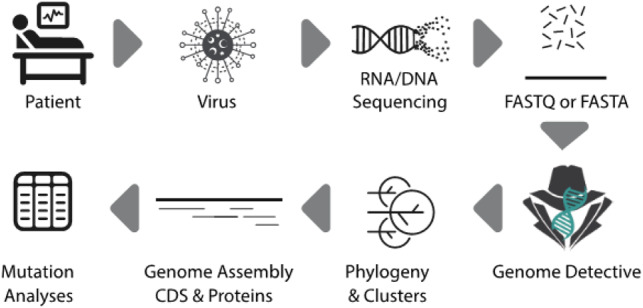
Summary: Genome detective is a web-based, user-friendly software application to quickly and accurately assemble all known virus genomes from next-generation sequencing datasets. This application allows the identification of phylogenetic clusters and genotypes from assembled genomes in FASTA format. Since its release in 2019, we have produced a number of typing tools for emergent viruses that have caused large outbreaks, such as Zika and Yellow Fever Virus in Brazil. Here, we present the Genome Detective Coronavirus Typing Tool that can accurately identify the novel severe acute respiratory syndrome (SARS)-related coronavirus (SARS-CoV-2) sequences isolated in China and around the world. The tool can accept up to 2000 sequences per submission and the analysis of a new whole-genome sequence will take approximately 1 min. The tool has been tested and validated with hundreds of whole genomes from 10 coronavirus species, and correctly classified all of the SARS-related coronavirus (SARSr-CoV) and all of the available public data for SARS-CoV-2. The tool also allows tracking of new viral mutations as the outbreak expands globally, which may help to accelerate the development of novel diagnostics, drugs and vaccines to stop the COVID-19 disease.
Availability and implementation: https://www.genomedetective.com/app/typingtool/cov.
Contact: koen@emweb.be or deoliveira@ukzn.ac.za.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press.
Figures
Update of
-
Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes.bioRxiv [Preprint]. 2020 Feb 2:2020.01.31.928796. doi: 10.1101/2020.01.31.928796. bioRxiv. 2020. Update in: Bioinformatics. 2020 Jun 1;36(11):3552-3555. doi: 10.1093/bioinformatics/btaa145. PMID: 32511309 Free PMC article. Updated. Preprint.
References
-
- Deforche K. (2017) An alignment method for nucleic acid sequences against annotated genomes. Biorxiv. doi:10.1101/200394.
-
- Gotoh O. (1982) An improved algorithm for matching biological sequences. J. Mol. Biol., 162, 705–708. - PubMed
-
- Guindon S., Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol., 52, 696–704. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
