

Polygenic Architecture of Human Neuroanatomical Diversity
- PMID: 32109272
- PMCID: PMC7175006
- DOI: 10.1093/cercor/bhz241
Polygenic Architecture of Human Neuroanatomical Diversity
Erratum in
-
Corrigendum: Polygenic Architecture of Human Neuroanatomical Diversity.Cereb Cortex. 2020 May 14;30(5):3435-3436. doi: 10.1093/cercor/bhaa093. Cereb Cortex. 2020. PMID: 32249901 Free PMC article. No abstract available.
Abstract
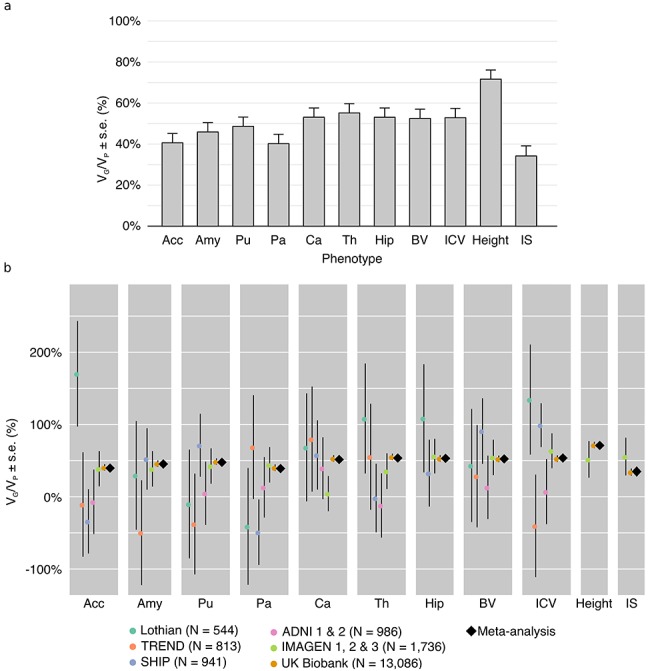
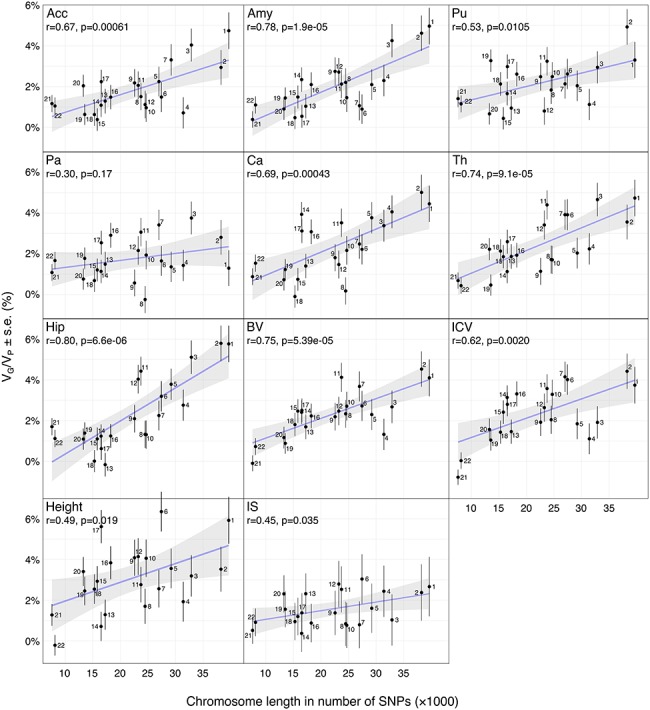
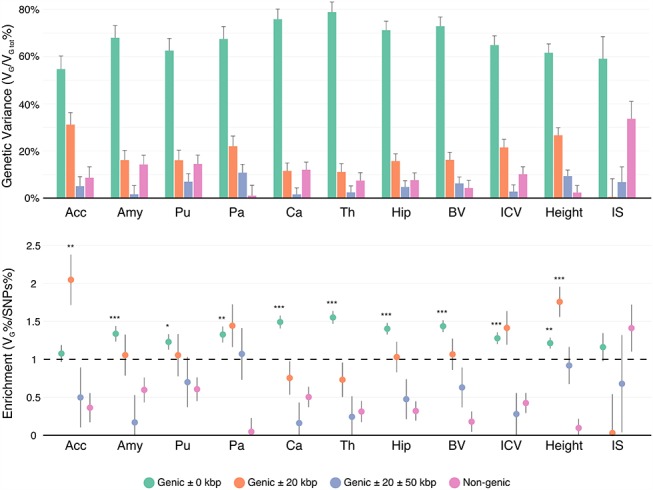
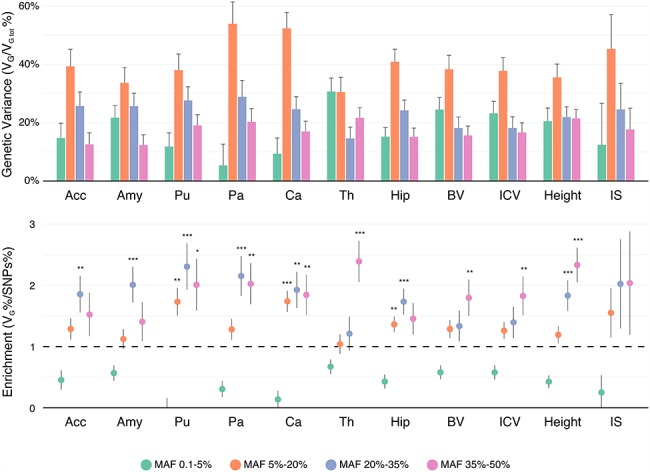
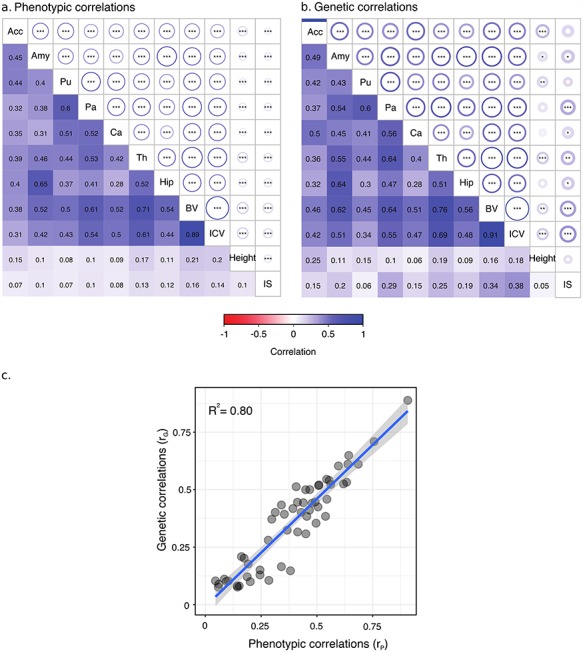
We analyzed the genomic architecture of neuroanatomical diversity using magnetic resonance imaging and single nucleotide polymorphism (SNP) data from >26 000 individuals from the UK Biobank project and 5 other projects that had previously participated in the ENIGMA (Enhancing NeuroImaging Genetics through Meta-Analysis) consortium. Our results confirm the polygenic architecture of neuroanatomical diversity, with SNPs capturing from 40% to 54% of regional brain volume variance. Chromosomal length correlated with the amount of phenotypic variance captured, r ~ 0.64 on average, suggesting that at a global scale causal variants are homogeneously distributed across the genome. At a local scale, SNPs within genes (~51%) captured ~1.5 times more genetic variance than the rest, and SNPs with low minor allele frequency (MAF) captured less variance than the rest: the 40% of SNPs with MAF <5% captured <one fourth of the genetic variance. We also observed extensive pleiotropy across regions, with an average genetic correlation of rG ~ 0.45. Genetic correlations were similar to phenotypic and environmental correlations; however, genetic correlations were often larger than phenotypic correlations for the left/right volumes of the same region. The heritability of differences in left/right volumes was generally not statistically significant, suggesting an important influence of environmental causes in the variability of brain asymmetry. Our code is available athttps://github.com/neuroanatomy/genomic-architecture.
Keywords: genetics; heritability; neuroimaging; polygenic architecture; subcortical structures.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JLR, Griffanti L, Douaud G, Sotiropoulos SN, Jbabdi S, Hernandez-Fernandez M, Vallee E et al. . 2018. Image processing and quality control for the first 10,000 brain imaging datasets from UK biobank. Neuroimage. 166(Feb):400–424. doi: 10.1016/j.neuroimage.2017.10.034. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
