

Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders
- PMID: 32109419
- PMCID: PMC7058823
- DOI: 10.1016/j.ajhg.2020.01.018
Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders
Abstract
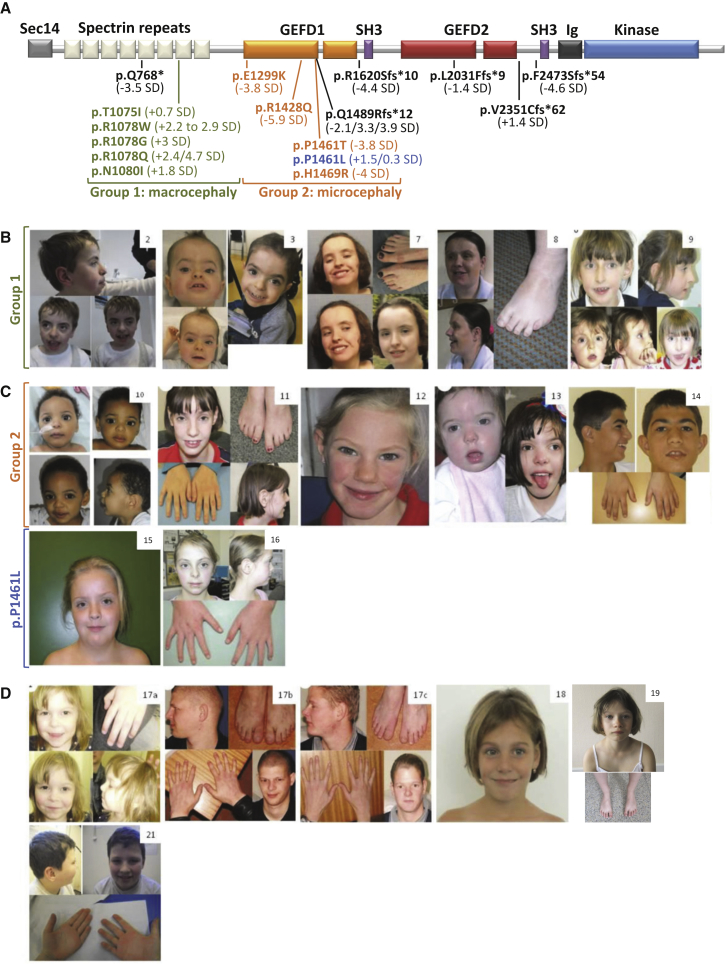
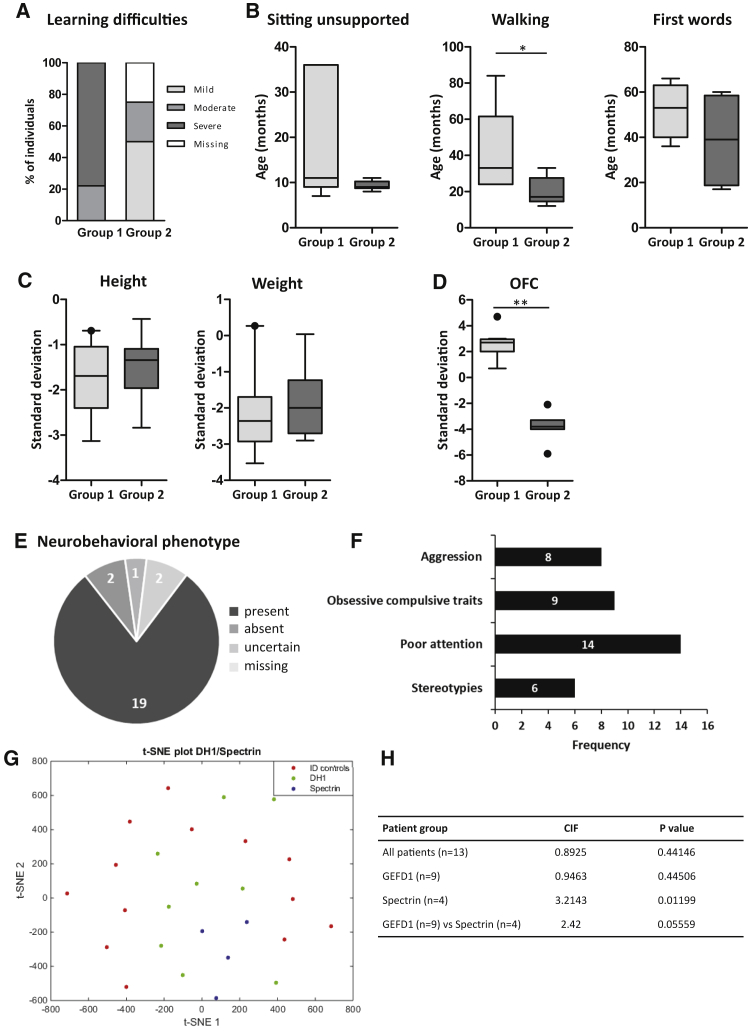
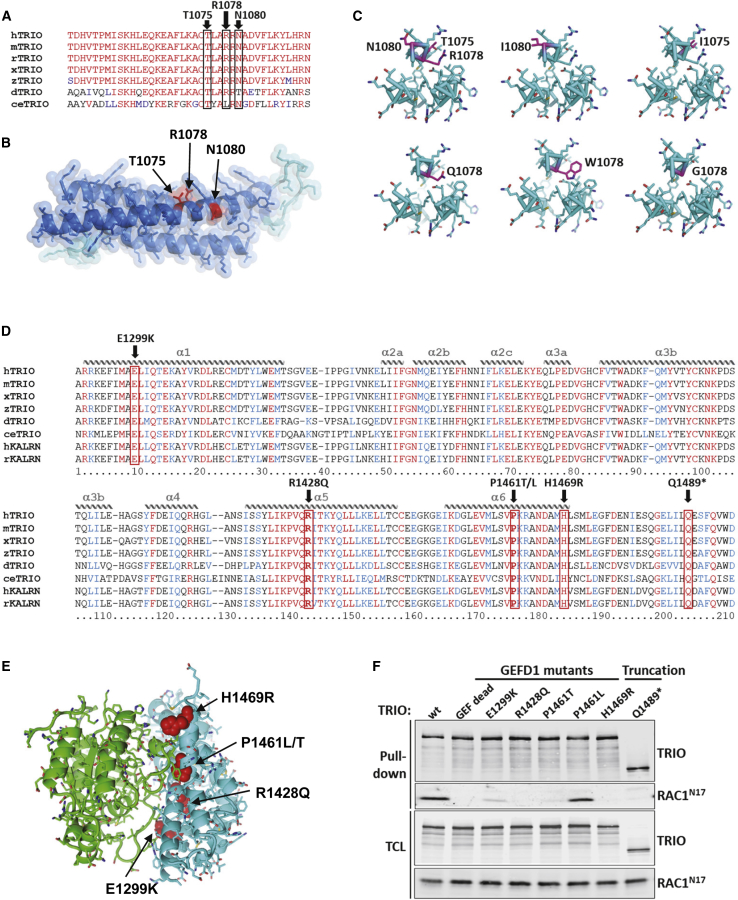
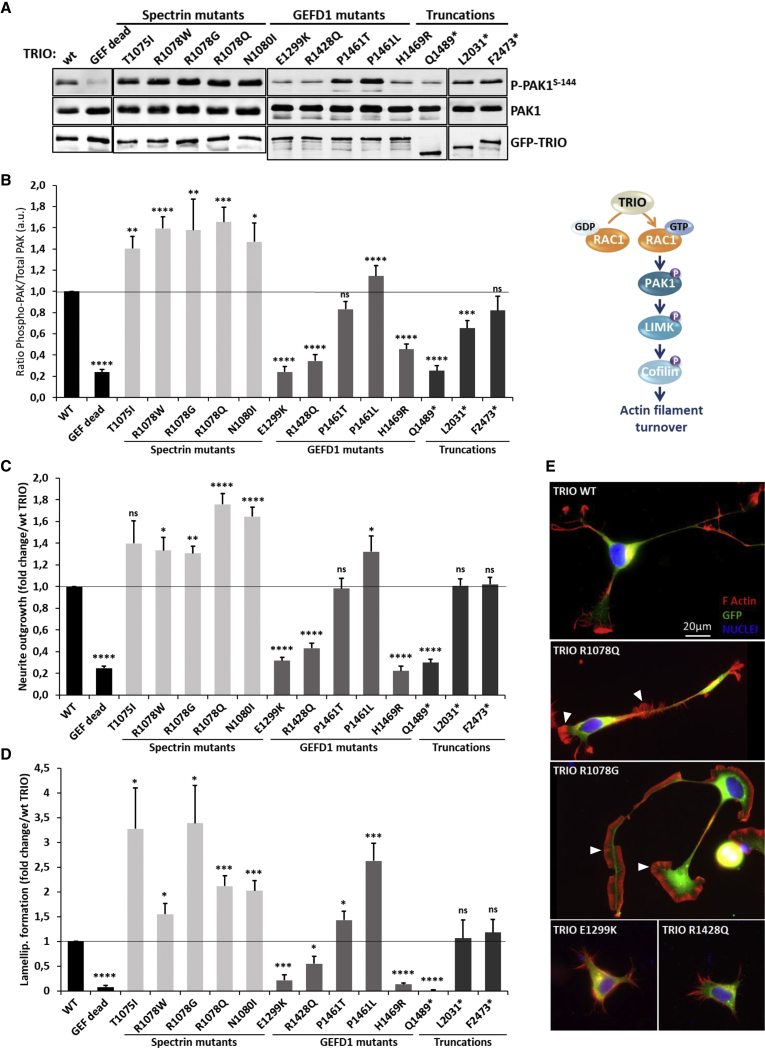
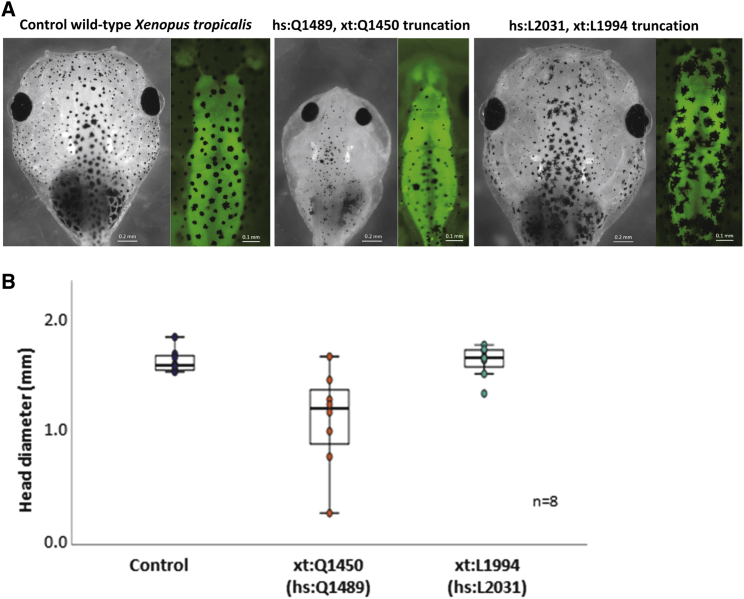
The Rho-guanine nucleotide exchange factor (RhoGEF) TRIO acts as a key regulator of neuronal migration, axonal outgrowth, axon guidance, and synaptogenesis by activating the GTPase RAC1 and modulating actin cytoskeleton remodeling. Pathogenic variants in TRIO are associated with neurodevelopmental diseases, including intellectual disability (ID) and autism spectrum disorders (ASD). Here, we report the largest international cohort of 24 individuals with confirmed pathogenic missense or nonsense variants in TRIO. The nonsense mutations are spread along the TRIO sequence, and affected individuals show variable neurodevelopmental phenotypes. In contrast, missense variants cluster into two mutational hotspots in the TRIO sequence, one in the seventh spectrin repeat and one in the RAC1-activating GEFD1. Although all individuals in this cohort present with developmental delay and a neuro-behavioral phenotype, individuals with a pathogenic variant in the seventh spectrin repeat have a more severe ID associated with macrocephaly than do most individuals with GEFD1 variants, who display milder ID and microcephaly. Functional studies show that the spectrin and GEFD1 variants cause a TRIO-mediated hyper- or hypo-activation of RAC1, respectively, and we observe a striking correlation between RAC1 activation levels and the head size of the affected individuals. In addition, truncations in TRIO GEFD1 in the vertebrate model X. tropicalis induce defects that are concordant with the human phenotype. This work demonstrates distinct clinical and molecular disorders clustering in the GEFD1 and seventh spectrin repeat domains and highlights the importance of tight control of TRIO-RAC1 signaling in neuronal development.
Keywords: autism; intellectual disability; macrocephaly; microcephaly.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Govek E.E., Newey S.E., Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. - PubMed
-
- Debant A., Serra-Pagès C., Seipel K., O’Brien S., Tang M., Park S.H., Streuli M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA. 1996;93:5466–5471. - PMC - PubMed
-
- Blangy A., Vignal E., Schmidt S., Debant A., Gauthier-Rouvière C., Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 2000;113:729–739. - PubMed
-
- Bellanger J.M., Lazaro J.B., Diriong S., Fernandez A., Lamb N., Debant A. The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene. 1998;16:147–152. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
