

Comparison of pathway and gene-level models for cancer prognosis prediction
- PMID: 32111152
- PMCID: PMC7048092
- DOI: 10.1186/s12859-020-3423-z
Comparison of pathway and gene-level models for cancer prognosis prediction
Abstract
Background: Cancer prognosis prediction is valuable for patients and clinicians because it allows them to appropriately manage care. A promising direction for improving the performance and interpretation of expression-based predictive models involves the aggregation of gene-level data into biological pathways. While many studies have used pathway-level predictors for cancer survival analysis, a comprehensive comparison of pathway-level and gene-level prognostic models has not been performed. To address this gap, we characterized the performance of penalized Cox proportional hazard models built using either pathway- or gene-level predictors for the cancers profiled in The Cancer Genome Atlas (TCGA) and pathways from the Molecular Signatures Database (MSigDB).
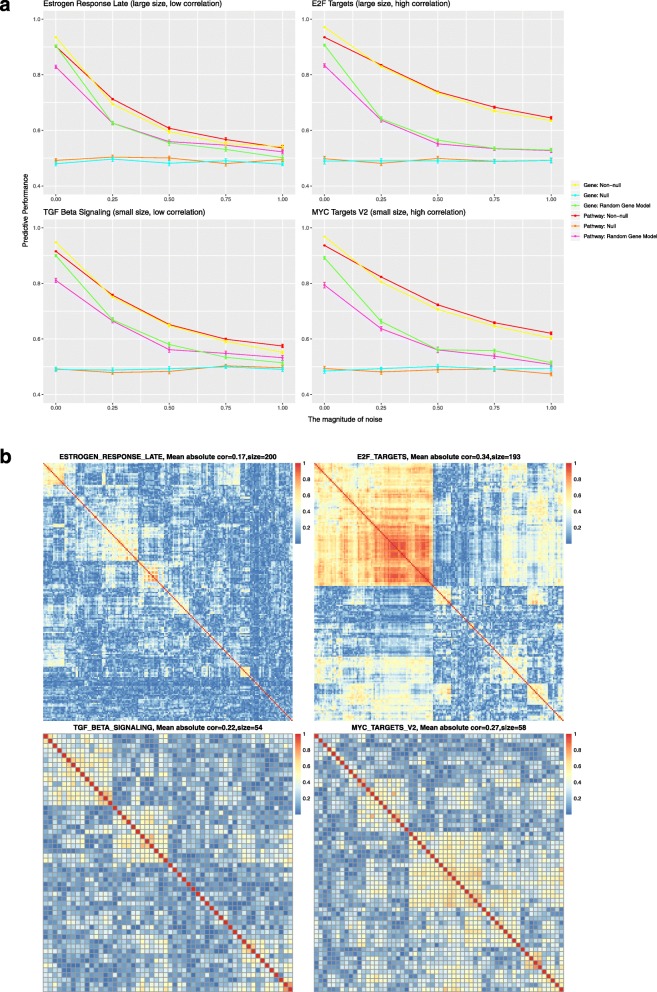
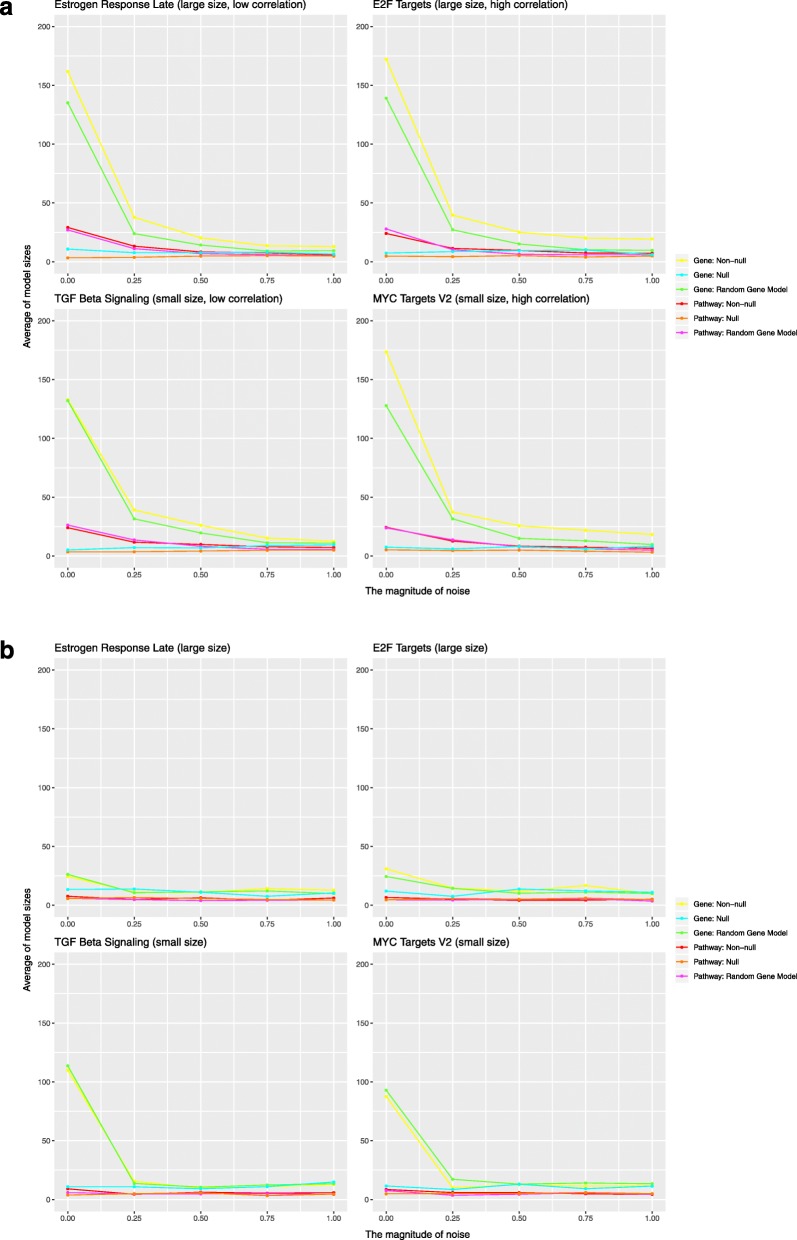
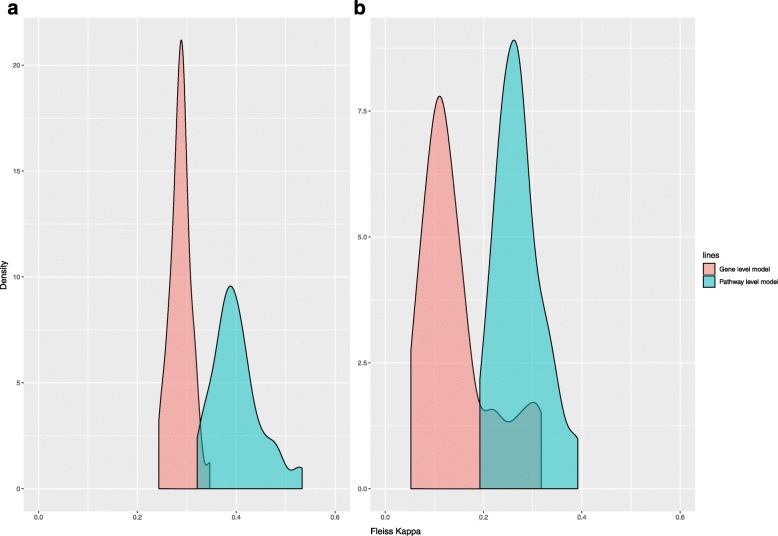
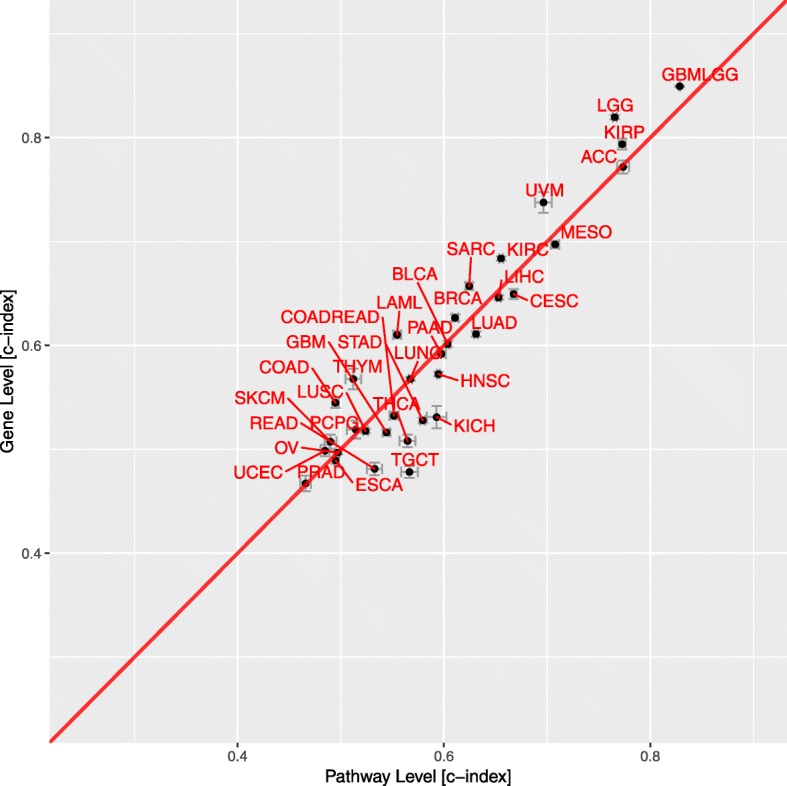
Results: When analyzing TCGA data, we found that pathway-level models are more parsimonious, more robust, more computationally efficient and easier to interpret than gene-level models with similar predictive performance. For example, both pathway-level and gene-level models have an average Cox concordance index of ~ 0.85 for the TCGA glioma cohort, however, the gene-level model has twice as many predictors on average, the predictor composition is less stable across cross-validation folds and estimation takes 40 times as long as compared to the pathway-level model. When the complex correlation structure of the data is broken by permutation, the pathway-level model has greater predictive performance while still retaining superior interpretative power, robustness, parsimony and computational efficiency relative to the gene-level models. For example, the average concordance index of the pathway-level model increases to 0.88 while the gene-level model falls to 0.56 for the TCGA glioma cohort using survival times simulated from uncorrelated gene expression data.
Conclusion: The results of this study show that when the correlations among gene expression values are low, pathway-level analyses can yield better predictive performance, greater interpretative power, more robust models and less computational cost relative to a gene-level model. When correlations among genes are high, a pathway-level analysis provides equivalent predictive power compared to a gene-level analysis while retaining the advantages of interpretability, robustness and computational efficiency.
Keywords: Cancer prognosis prediction; Gene expression data; Inter-gene correlation; L1 penalized regression model; Pathway analysis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Barillot E. Computational systems biology of Cancer. Boca Raton: CRC Press; 2012.
-
- Tandon AK, Clark GM, Chamness GC, Ullrich A, McGuire WL. HER-2/neu oncogene protein and prognosis in breast cancer. J Clin Oncol. 1989;7(8):1120–1128. - PubMed
-
- Jenssen TK, Kuo WP, Stokke T, Hovig E. Associations between gene expressions in breast cancer and patient survival. Hum Genet. 2002;111(4–5):411–420. - PubMed
-
- Park MY, Hastie T. L1-regularization path algorithm for generalized linear models. J R Stat Soc Ser B Stat Methodol. 2007;69(4):659–677.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
