

Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China
- PMID: 32111191
- PMCID: PMC7049201
- DOI: 10.1186/s12881-020-0981-x
Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China
Abstract
Background: Individuals with δβ-thalassemia/HPFH and β-thalassemia usually present with intermedia or thalassemia major. No large-scale survey on HPFH/δβ-thalassemia in southern China has been reported to date. The purpose of this study was to examine the molecular epidemiology and hematologic characteristics of these disorders in Guangzhou, the largest city in Southern China, to offer advice for thalassemia screening programs and genetic counseling.
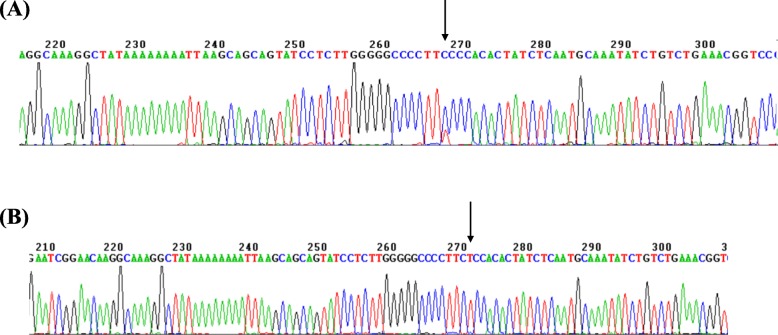
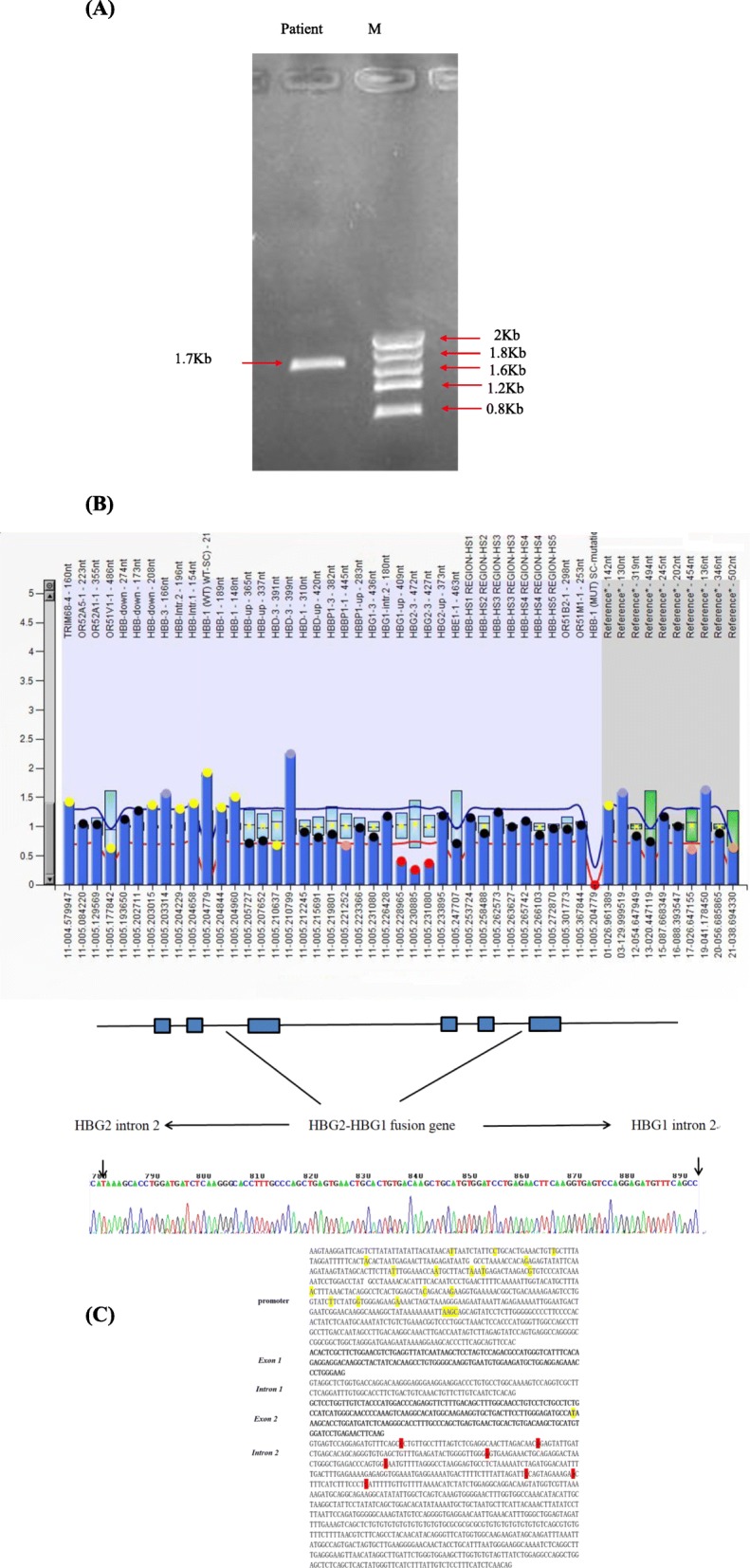
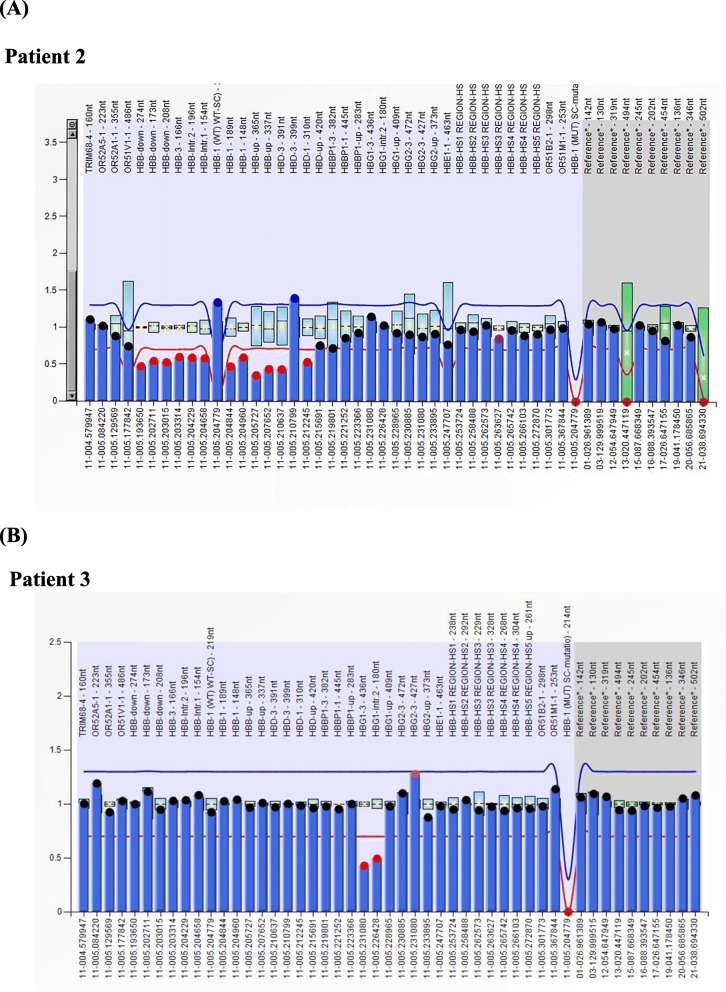
Methods: A total of 125,661 couples participated in pregestational thalassemia screening. 654 subjects with fetal hemoglobin (HbF) level ≥ 5% were selected for further investigation. Gap-PCR combined with Multiplex ligation dependent probe amplification (MLPA) was used to screen for β-globin gene cluster deletions. Gene sequencing for the promoter region of HBG1 /HBG2 gene was performed for all those subjects.
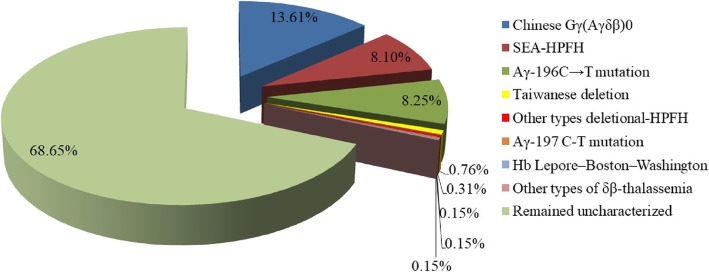
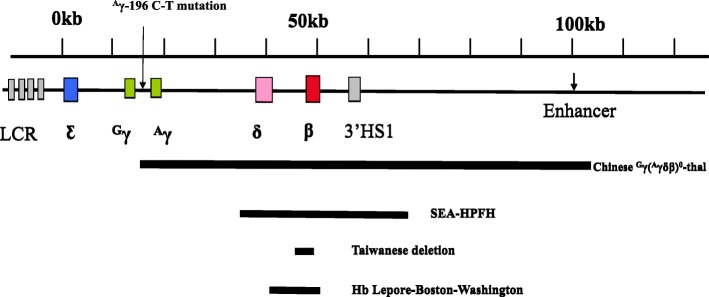
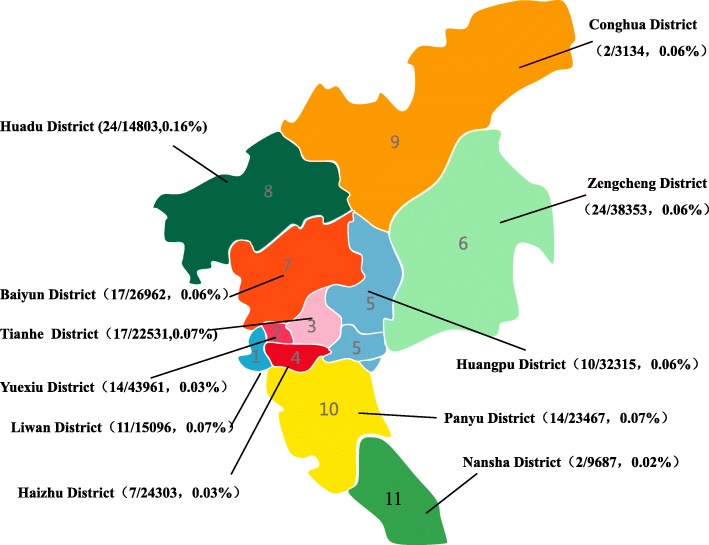
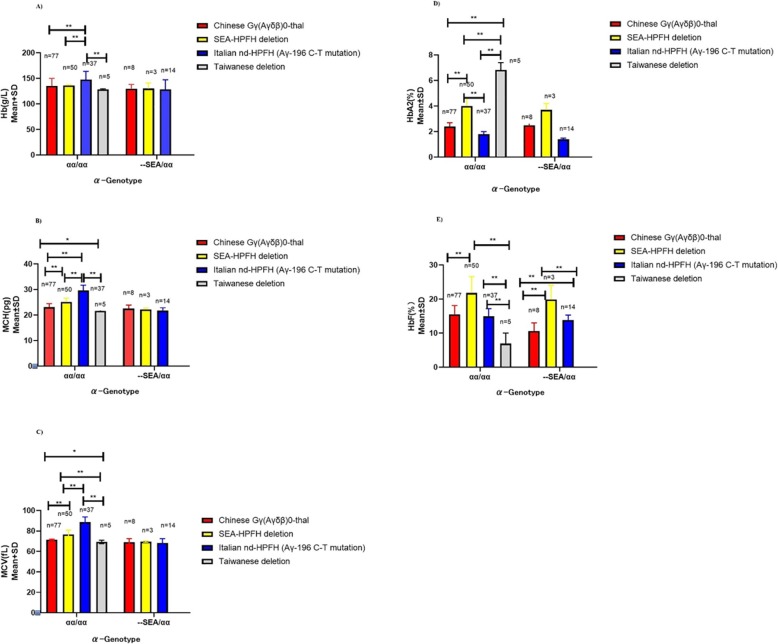
Results: A total of 654 individuals had hemoglobin (HbF) levels≥5, and 0.12% of the couples were found to be heterozygous for HPFH/δβ-thalassemia, including Chinese Gγ (Aγδβ)0-thal, Southeast Asia HPFH (SEA-HPFH), Taiwanese deletion and Hb Lepore-Boston-Washington. The highest prevalence was observed in the Huadu district and the lowest in the Nansha district. Three cases were identified as carrying β-globin gene cluster deletions, which had not been previously reported. Two at-risk couples (0.0015%) were required to receive prenatal diagnosis. We also found 55cases of nondeletional-HPFH (nd-HPFH), including 54 with Italian nd-HPFH and one with the Aγ-197C-T heterozygous state. It is difficult to discriminate between Chinese Gγ (Aγδβ)0-thal and Italian nd-HPFH carriers using hemoglobin (Hb) analysis.
Conclusions: This study is the first to describe the familial prevalence of HPFH/δβ-thalassemia and the high-risk rate in Greater Guangzhou Area, and the findings will support the implementation of thalassemia screening for three common deletions by gap-PCR. We also presented a systematic description of genotype-phenotype relationships which will be useful for genetic counseling and prenatal diagnostic services for β-thalassemia intermedia.
Keywords: Guangzhou; HPFH; Prevalence; δβ-thalassemia.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
The prevalence and molecular characterization of (δβ)0 -thalassemia and hereditary persistence of fetal hemoglobin in the Chinese Zhuang population.J Clin Lab Anal. 2018 Mar;32(3):e22304. doi: 10.1002/jcla.22304. Epub 2017 Aug 1. J Clin Lab Anal. 2018. PMID: 28763119 Free PMC article.
-
Molecular Characterization of δβ Thalassemia/Hereditary Persistence of Fetal Hemoglobin and Its Correlation With Clinical and Hematological Profile; a Single Center Study in North India.Int J Lab Hematol. 2025 Apr;47(2):318-325. doi: 10.1111/ijlh.14419. Epub 2024 Dec 27. Int J Lab Hematol. 2025. PMID: 39731309
-
Genetic research and clinical analysis of β-globin gene cluster deletions in the Chinese population of Fujian province: A 14-year single-center experience.J Clin Lab Anal. 2022 Feb;36(2):e24181. doi: 10.1002/jcla.24181. Epub 2021 Dec 23. J Clin Lab Anal. 2022. PMID: 34951062 Free PMC article.
-
Interpreting elevated fetal hemoglobin in pathology and health at the basic laboratory level: new and known γ- gene mutations associated with hereditary persistence of fetal hemoglobin.Int J Lab Hematol. 2014 Feb;36(1):13-9. doi: 10.1111/ijlh.12094. Epub 2013 Apr 29. Int J Lab Hematol. 2014. PMID: 23621512 Review.
-
Molecular basis of hereditary persistence of fetal hemoglobin.Ann N Y Acad Sci. 1998 Jun 30;850:38-44. doi: 10.1111/j.1749-6632.1998.tb10460.x. Ann N Y Acad Sci. 1998. PMID: 9668525 Review.
Cited by
-
Analysis of rare thalassemia genetic variants based on third-generation sequencing.Sci Rep. 2022 Jun 14;12(1):9907. doi: 10.1038/s41598-022-14038-8. Sci Rep. 2022. PMID: 35701592 Free PMC article.
-
Long-Read Sequencing Identified a Large Novel δ/β-Globin Gene Deletion in a Chinese Family.Hum Mutat. 2023 Oct 4;2023:2766625. doi: 10.1155/2023/2766625. eCollection 2023. Hum Mutat. 2023. PMID: 40225154 Free PMC article.
-
Utilization of multiple genetic methods for prenatal diagnosis of rare thalassemia variants.Front Genet. 2023 Jul 17;14:1208102. doi: 10.3389/fgene.2023.1208102. eCollection 2023. Front Genet. 2023. PMID: 37529778 Free PMC article.
-
The 1357 bp deletion in β-thalassemia: molecular profiling and hematological characterization in a Guangxi cohort.Mol Biol Rep. 2025 Jun 16;52(1):602. doi: 10.1007/s11033-025-10724-8. Mol Biol Rep. 2025. PMID: 40522534
-
Carrier Screening Programs for Cystic Fibrosis, Fragile X Syndrome, Hemoglobinopathies and Thalassemia, and Spinal Muscular Atrophy: A Health Technology Assessment.Ont Health Technol Assess Ser. 2023 Aug 10;23(4):1-398. eCollection 2023. Ont Health Technol Assess Ser. 2023. PMID: 37637488 Free PMC article.
References
-
- DJ W, JB C. The thalassemia syndromes. 4. Oxford: Blackwell Scientific; 2001.
-
- Bilgen T, Altiok Clark O, Ozturk Z, Yesilipek MA, Keser I. Gap-PCR screening for common large Deletional mutations of beta-globin gene cluster revealed a higher prevalence of the Turkish inversion/deletion (deltabeta)0 mutation in Antalya. Turk J Haematol. 2016;33(2):107–111. doi: 10.4274/tjh.2014.0242. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
