

Distribution and genetic diversity of adeno-associated viruses in bats from coastal areas of Southeast China
- PMID: 32111911
- PMCID: PMC7048818
- DOI: 10.1038/s41598-020-60721-z
Distribution and genetic diversity of adeno-associated viruses in bats from coastal areas of Southeast China
Abstract
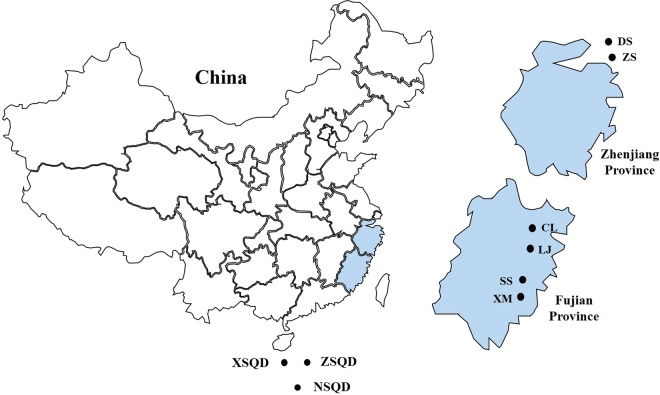
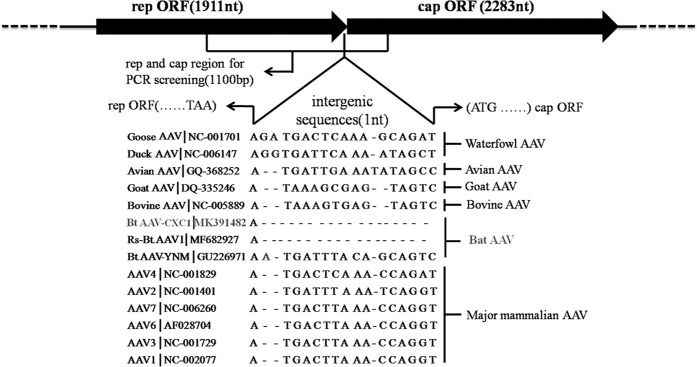
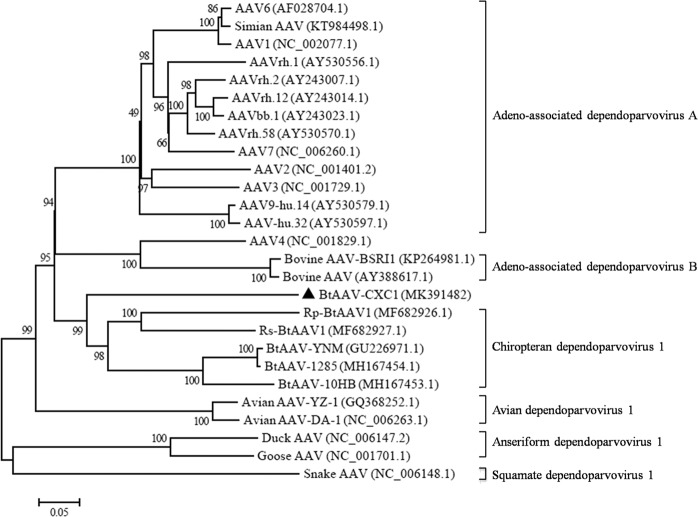
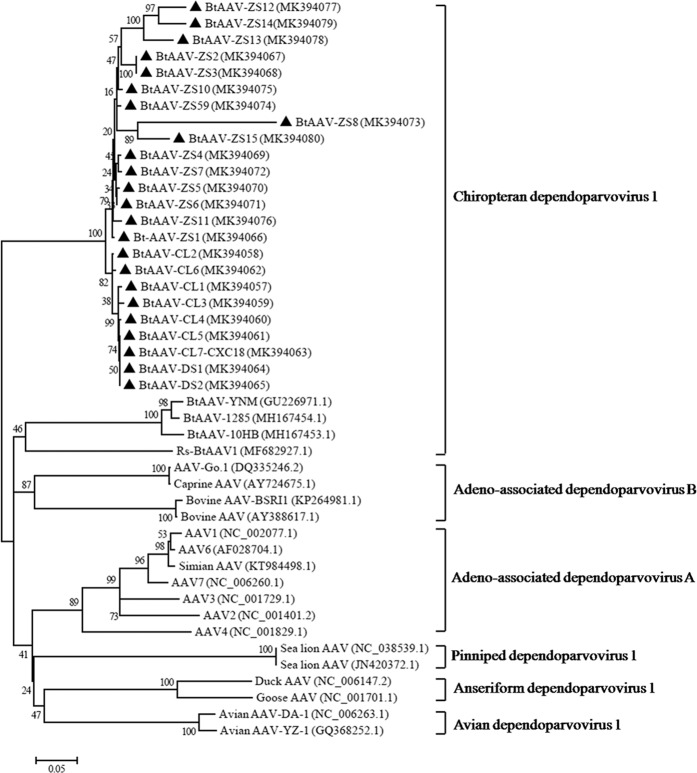
Bats are associated with several important zoonotic viruses from different families. One example includes adeno-associated viruses (AAVs), that are extensively detected in several animals, especially primates. To understand AAVs distribution and genetic diversity in the coastal areas of Southeast China, a total of 415 intestine samples were mostly collected from two provinces of southeast China, i.e., Zhejiang and Fujian province. Intestine samples from five bat species were collected for AAVs detection. The average prevalence rate for AAV detection among these samples was 18.6% (77 positives out of 415 samples) and ranged from 11.8 to 28.9% between the five bat species. This suggests that AAVs are widely distributed in diverse bat populations in southeast coastal areas of China. Based on the genome sequence of bat adeno-associated virus-CXC1(BtAAV-CXC1) from one AAV-positive sample, the genetic diversity of the detected AAVs were assessed and analyzed. Phylogenetic analysis revealed that BtAAV-CXC1 was comparatively distant to other major AAVs from mammals and non-mammals, with only a 52.9~64.7% nucleotide identity. However, they were phylogenetically closer to Rhinolophus sinicus bat adeno-associated virus (Rs-BtAAV1), with a 74.5% nt similarity. Partial analysis of the rep and cap overlapping open reading frame (ORF) sequences from bat AAV samples revealed 48 partial rep sequences and 23 partial cap sequences from positive samples shared 86.9 to 100% and 72.3 to 98.8% nucleotide identities among themselves, respectively. This suggests that the detected AAVs had a distinctly high genetic diversity. These findings led us to conclude that diverse AAVs may be widely distributed in bat populations from the southeast regions of China.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Prevalence and genetic diversity of adeno-associated viruses in bats from China.J Gen Virol. 2010 Oct;91(Pt 10):2601-9. doi: 10.1099/vir.0.020032-0. Epub 2010 Jun 23. J Gen Virol. 2010. PMID: 20573859
-
Novel Bat Alphacoronaviruses in Southern China Support Chinese Horseshoe Bats as an Important Reservoir for Potential Novel Coronaviruses.Viruses. 2019 May 7;11(5):423. doi: 10.3390/v11050423. Viruses. 2019. PMID: 31067830 Free PMC article.
-
Bats host diverse parvoviruses as possible origin of mammalian dependoparvoviruses and source for bat-swine interspecies transmission.J Gen Virol. 2017 Dec;98(12):3046-3059. doi: 10.1099/jgv.0.000969. J Gen Virol. 2017. PMID: 29106348
-
Identification and interspecies transmission of a novel bocaparvovirus among different bat species in China.J Gen Virol. 2016 Dec;97(12):3345-3358. doi: 10.1099/jgv.0.000645. Epub 2016 Oct 25. J Gen Virol. 2016. PMID: 27902362
-
Virome analysis for identification of novel mammalian viruses in bats from Southeast China.Sci Rep. 2017 Sep 7;7(1):10917. doi: 10.1038/s41598-017-11384-w. Sci Rep. 2017. PMID: 28883450 Free PMC article.
Cited by
-
Small but mighty: old and new parvoviruses of veterinary significance.Virol J. 2021 Oct 24;18(1):210. doi: 10.1186/s12985-021-01677-y. Virol J. 2021. PMID: 34689822 Free PMC article. Review.
-
Integration of adeno-associated virus (AAV) into the genomes of most Thai and Mongolian liver cancer patients does not induce oncogenesis.BMC Genomics. 2021 Nov 11;22(1):814. doi: 10.1186/s12864-021-08098-9. BMC Genomics. 2021. PMID: 34763675 Free PMC article.
-
Specific detection of duck adeno-associated virus using a TaqMan-based real-time PCR assay.Front Vet Sci. 2024 Nov 13;11:1483990. doi: 10.3389/fvets.2024.1483990. eCollection 2024. Front Vet Sci. 2024. PMID: 39606664 Free PMC article.
-
Adeno-Associated Virus (AAV) Gene Delivery: Dissecting Molecular Interactions upon Cell Entry.Viruses. 2021 Jul 10;13(7):1336. doi: 10.3390/v13071336. Viruses. 2021. PMID: 34372542 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
