

Zebrafish as a Model System for the Study of Severe CaV2.1 (α1A) Channelopathies
- PMID: 32116539
- PMCID: PMC7018710
- DOI: 10.3389/fnmol.2019.00329
Zebrafish as a Model System for the Study of Severe CaV2.1 (α1A) Channelopathies
Abstract
The P/Q-type CaV2.1 channel regulates neurotransmitter release at neuromuscular junctions (NMJ) and many central synapses. CACNA1A encodes the pore-containing α1A subunit of CaV2.1 channels. In humans, de novo CACNA1A mutations result in a wide spectrum of neurological, neuromuscular, and movement disorders, such as familial hemiplegic migraine type 1 (FHM1), episodic ataxia type 2 (EA2), as well as a more recently discovered class of more severe disorders, which are characterized by ataxia, hypotonia, cerebellar atrophy, and cognitive/developmental delay. Heterologous expression of CaV2.1 channels has allowed for an understanding of the consequences of CACNA1A missense mutations on channel function. In contrast, a mechanistic understanding of how specific CACNA1A mutations lead in vivo to the resultant phenotypes is lacking. In this review, we present the zebrafish as a model to both study in vivo mechanisms of CACNA1A mutations that result in synaptic and behavioral defects and to screen for effective drug therapies to combat these and other CaV2.1 channelopathies.
Keywords: CaV2.1; P/Q-type; channelopathy; episodic ataxia type 2; familial hemiplegic migraine type 1; vertebrate models; zebrafish; α1A.
Copyright © 2020 Tyagi, Ribera and Bannister.
Figures
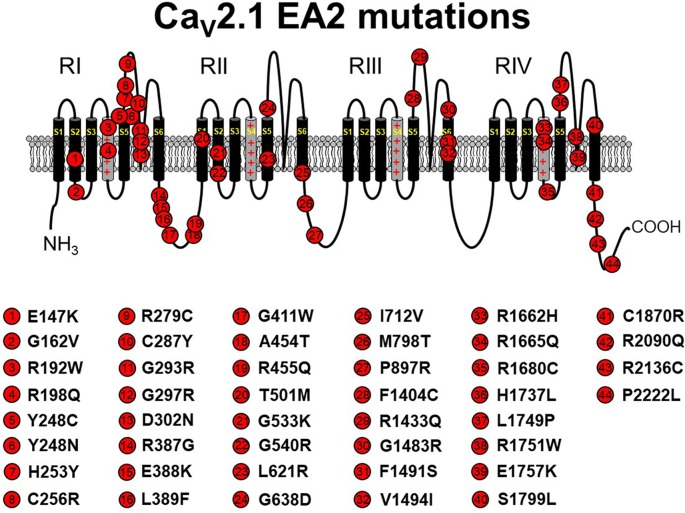
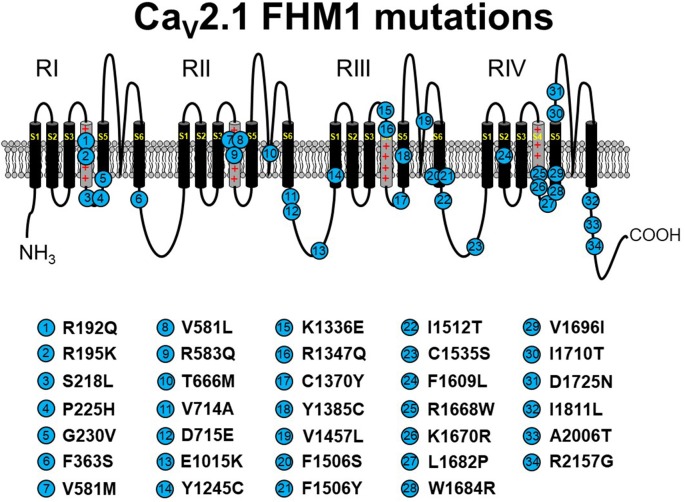
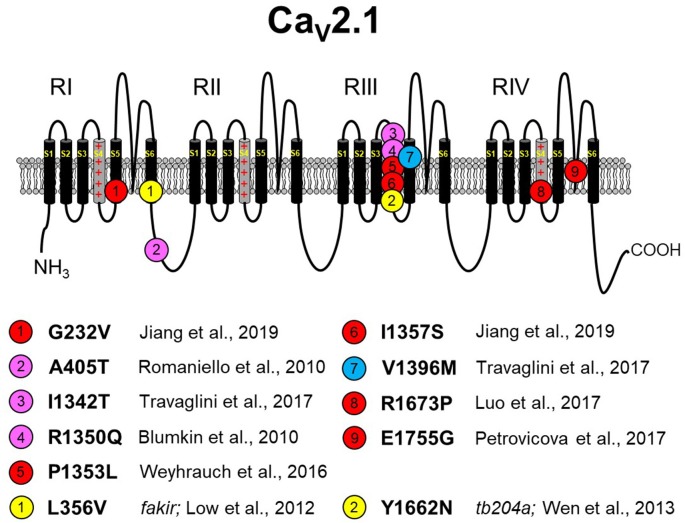
References
-
- Adams P. J., Garcia E., David L. S., Mulatz K. J., Spacey S. D., Snutch T. P. (2009). CaV2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels 3, 110–121. 10.4161/chan.3.2.7932 - DOI - PubMed
-
- Alonso I., Barros J., Tuna A., Seixas A., Coutinho P., Sequeiros J., et al. . (2004). A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel α-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine. Clin. Genet. 65, 70–72. 10.1111/j..2004.00187.x - DOI - PubMed
-
- Bahamonde M. I., Serra S. A., Drechsel O., Rahman R., Marcé-Grau A., Prieto M., et al. . (2015). A single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS One 10:e0146035. 10.1371/journal.pone.0146035 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources