

Evaluation of the Efficacy and Safety of Switching to Pasireotide in Patients With Acromegaly Inadequately Controlled With First-Generation Somatostatin Analogs
- PMID: 32117045
- PMCID: PMC7008501
- DOI: 10.3389/fendo.2019.00931
Evaluation of the Efficacy and Safety of Switching to Pasireotide in Patients With Acromegaly Inadequately Controlled With First-Generation Somatostatin Analogs
Abstract
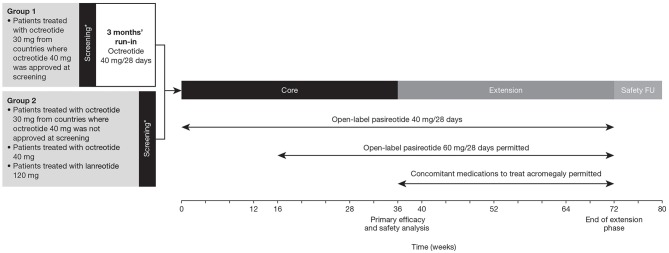
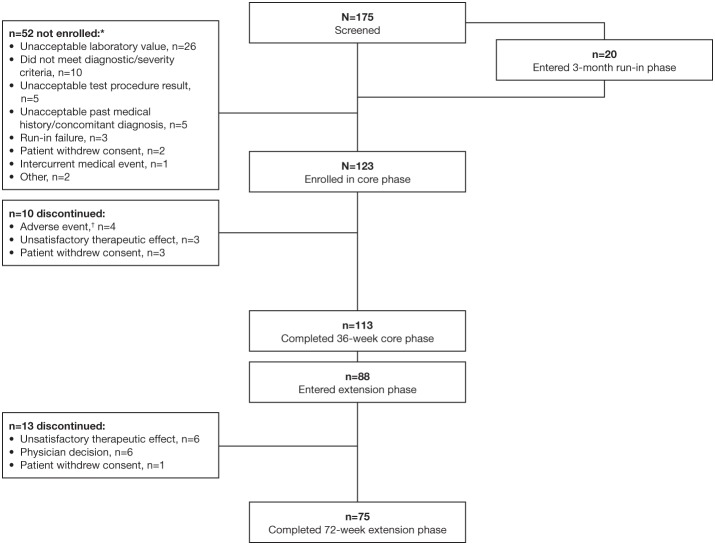
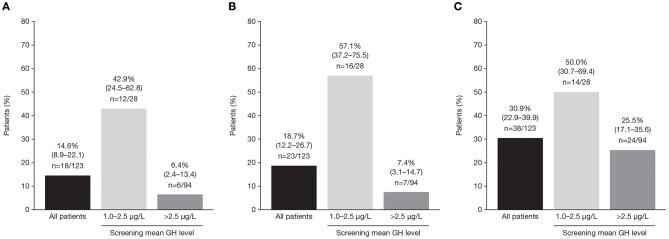
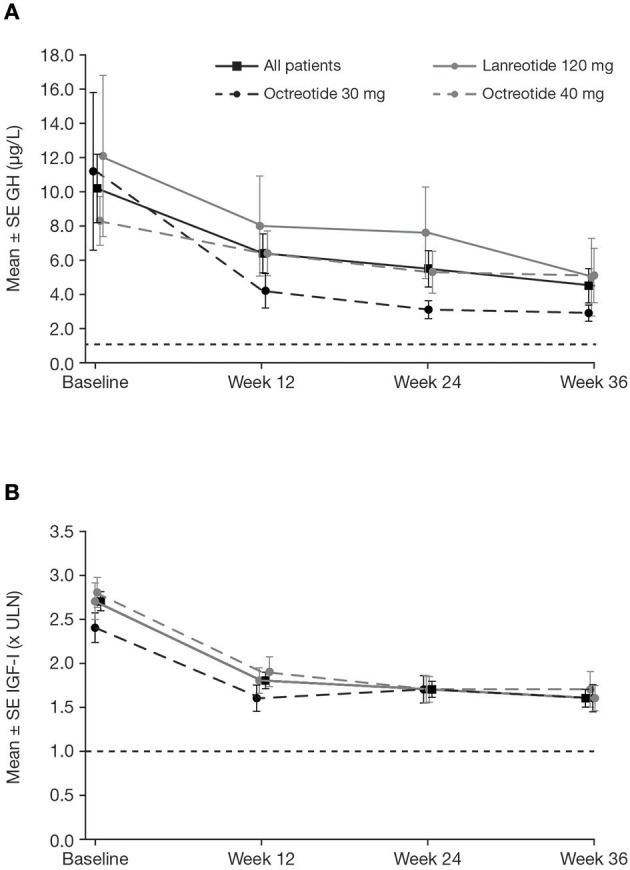
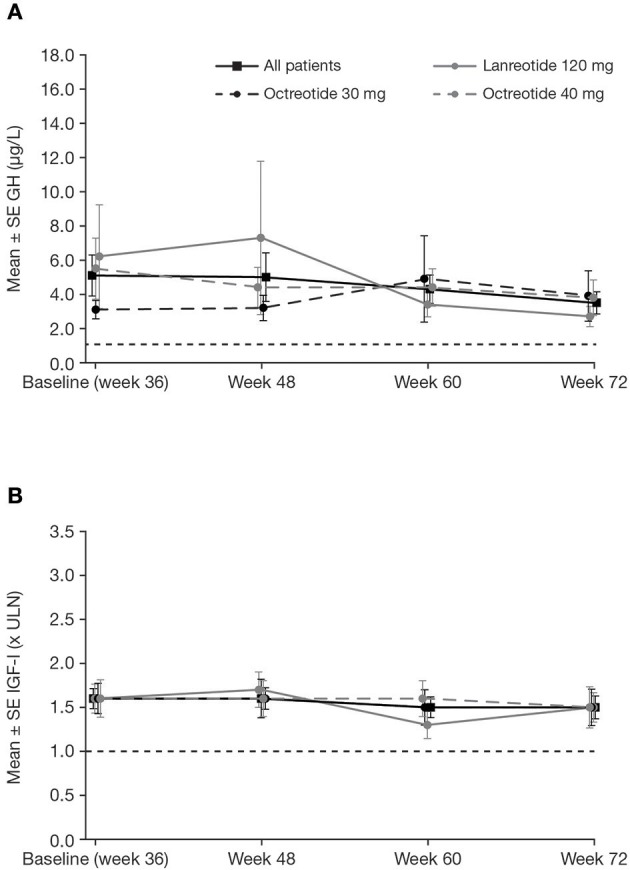
Introduction: Acromegaly is a rare, serious endocrine disorder characterized by excess growth hormone (GH) secretion by a pituitary adenoma and overproduction of insulin-like growth factor I (IGF-I). Transsphenoidal surgery is the treatment of choice, although many patients require additional interventions. First-generation somatostatin analogs (SSAs) are the current standard of medical therapy; however, not all patients achieve control of GH and IGF-I. Outcomes from a Phase IIIb open-label study of patients with uncontrolled acromegaly on first-generation SSAs switching to pasireotide are reported. Methods: Adults with uncontrolled acromegaly (mean GH [mGH] ≥1 μg/L from a five-point profile over 2 h, and IGF-I >1.3× upper limit of normal [ULN]) despite ≥3 months' treatment with maximal approved doses of long-acting octreotide/lanreotide received open-label long-acting pasireotide 40 mg/28 days. Pasireotide dose could be increased (maximum: 60 mg/28 days) after week 12 if biochemical control was not achieved, or decreased (minimum: 10 mg/28 days) for tolerability. Patients who completed 36 weeks' treatment could continue receiving pasireotide during an extension (weeks 36-72) when concomitant medication for acromegaly was permitted. Primary endpoint was proportion of patients with mGH <1 μg/L and IGF-I <ULN at week 36. Biochemical control was also assessed during the extension. Safety was assessed throughout. Results: One hundred and twenty-three patients were enrolled and received pasireotide; 88 patients continued into the extension. Overall, 18 [14.6% (95% CI: 8.9-22.1)] patients achieved mGH <1 μg/L and IGF-I <ULN at week 36; biochemical control was achieved in 42.9% with screening mGH 1.0-2.5 μg/L and 6.4% with screening mGH >2.5 μg/L. For patients who entered the extension, 14.8% (95% CI: 8.1-23.9), 12.5% (95% CI: 6.4-21.3), 14.8% (95% CI: 8.1-23.9) and 11.4% (95% CI: 5.6-19.9) had mGH <1 μg/L and IGF-I <ULN at weeks 36, 48, 60, and 72, respectively. During the overall study period, most frequent investigator-reported drug-related adverse events were hyperglycemia (41.5%), diabetes mellitus (23.6%), and diarrhea (11.4%). Conclusions: Switching to long-acting pasireotide provided biochemical control in some patients, which was sustained with continued treatment. Long-term safety and tolerability of long-acting pasireotide was consistent with the known safety profile. These data support long-acting pasireotide for some patients with acromegaly who are uncontrolled on first generation SSAs. Clinical Trial Registration: clinicaltrials.gov, identifier: NCT02354508.
Keywords: acromegaly; growth hormone; insulin-like growth factor I; pasireotide; somatostatin.
Copyright © 2020 Gadelha, Bex, Colao, Pedroza García, Poiana, Jimenez-Sanchez, Yener, Mukherjee, Bartalotta, Maamari and Raverot.
Figures
References
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical