

The Genetics and Epigenetics of 22q11.2 Deletion Syndrome
- PMID: 32117416
- PMCID: PMC7016268
- DOI: 10.3389/fgene.2019.01365
The Genetics and Epigenetics of 22q11.2 Deletion Syndrome
Abstract
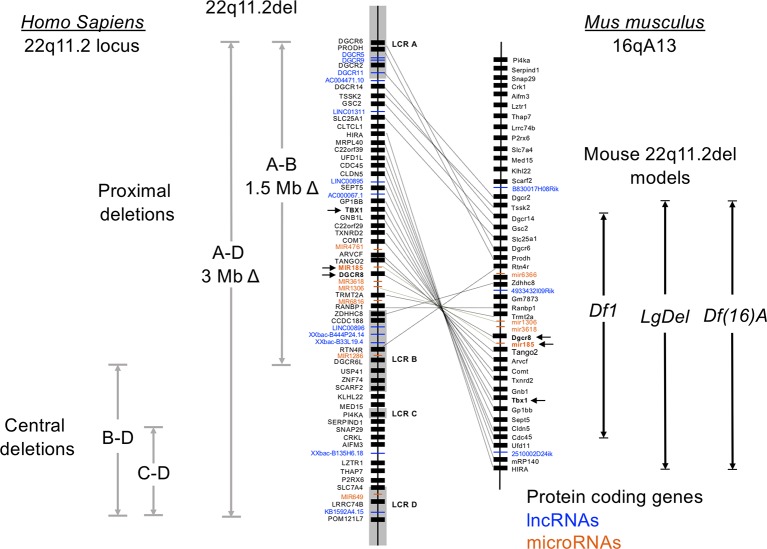
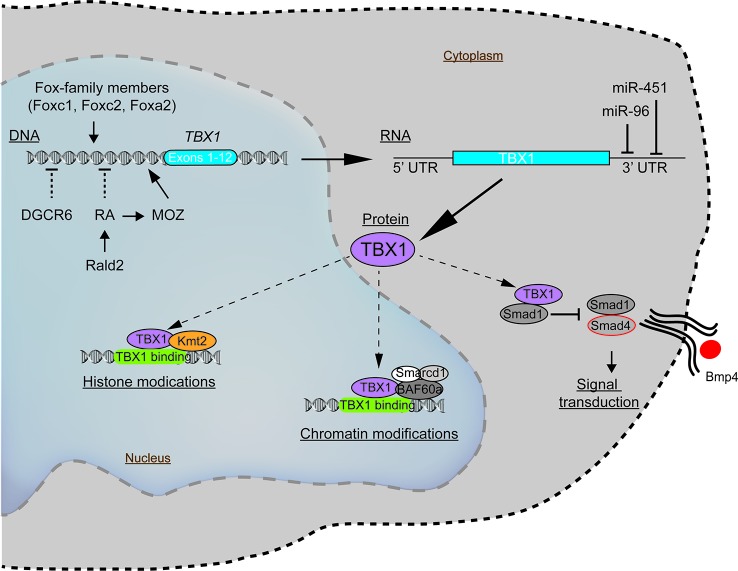
Chromosome 22q11.2 deletion syndrome (22q11.2del) is a complex, multi-organ disorder noted for its varying severity and penetrance among those affected. The clinical problems comprise congenital malformations; cardiac problems including outflow tract defects, hypoplasia of the thymus, hypoparathyroidism, and/or dysmorphic facial features. Additional clinical issues that can appear over time are autoimmunity, renal insufficiency, developmental delay, malignancy and neurological manifestations such as schizophrenia. The majority of individuals with 22q11.2del have a 3 Mb deletion of DNA on chromosome 22, leading to a haploinsufficiency of ~106 genes, which comprise coding RNAs, noncoding RNAs, and pseudogenes. The consequent haploinsufficiency of many of the coding genes are well described, including the key roles of T-box Transcription Factor 1 (TBX1) and DiGeorge Critical Region 8 (DGCR8) in the clinical phenotypes. However, the haploinsufficiency of these genes alone cannot account for the tremendous variation in the severity and penetrance of the clinical complications among those affected. Recent RNA and DNA sequencing approaches are uncovering novel genetic and epigenetic differences among 22q11.2del patients that can influence disease severity. In this review, the role of coding and non-coding genes, including microRNAs (miRNA) and long noncoding RNAs (lncRNAs), will be discussed in relation to their bearing on 22q11.2del with an emphasis on TBX1.
Keywords: 22q11.2 deletion syndrome; DiGeorge syndrome; TBX1; epigenetics; haploinsufficiency; microRNAs; noncoding RNAs.
Copyright © 2020 Du, de la Morena and van Oers.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous