

Relative Efficiencies of Simple and Complex Substitution Models in Estimating Divergence Times in Phylogenomics
- PMID: 32119075
- PMCID: PMC7253201
- DOI: 10.1093/molbev/msaa049
Relative Efficiencies of Simple and Complex Substitution Models in Estimating Divergence Times in Phylogenomics
Abstract
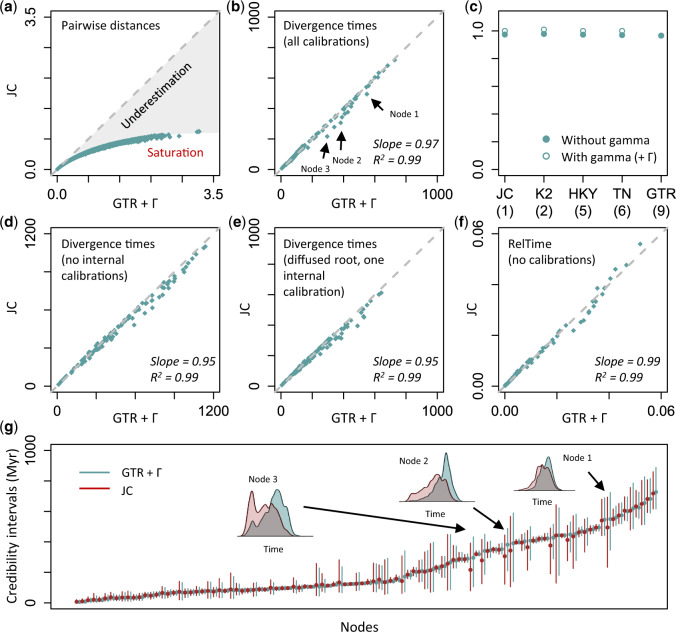
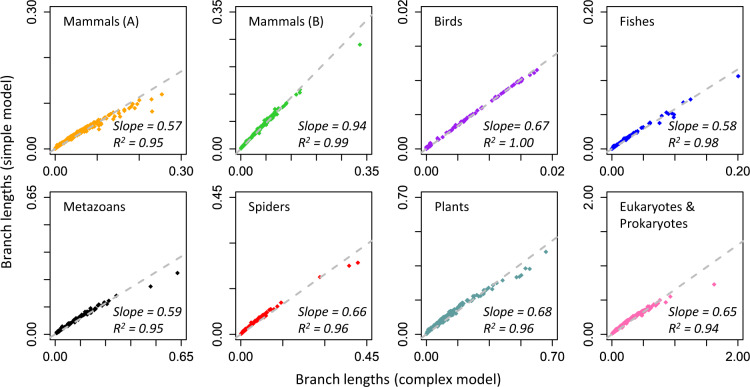
The conventional wisdom in molecular evolution is to apply parameter-rich models of nucleotide and amino acid substitutions for estimating divergence times. However, the actual extent of the difference between time estimates produced by highly complex models compared with those from simple models is yet to be quantified for contemporary data sets that frequently contain sequences from many species and genes. In a reanalysis of many large multispecies alignments from diverse groups of taxa, we found that the use of the simplest models can produce divergence time estimates and credibility intervals similar to those obtained from the complex models applied in the original studies. This result is surprising because the use of simple models underestimates sequence divergence for all the data sets analyzed. We found three fundamental reasons for the observed robustness of time estimates to model complexity in many practical data sets. First, the estimates of branch lengths and node-to-tip distances under the simplest model show an approximately linear relationship with those produced by using the most complex models applied on data sets with many sequences. Second, relaxed clock methods automatically adjust rates on branches that experience considerable underestimation of sequence divergences, resulting in time estimates that are similar to those from complex models. And, third, the inclusion of even a few good calibrations in an analysis can reduce the difference in time estimates from simple and complex models. The robustness of time estimates to model complexity in these empirical data analyses is encouraging, because all phylogenomics studies use statistical models that are oversimplified descriptions of actual evolutionary substitution processes.
Keywords: RelTime; molecular dating; phylogenomics; relaxed clock; substitution model.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures






References
-
- Alfaro ME, Faircloth BC, Harrington RC, Sorenson L, Friedman M, Thacker CE, Oliveros CH, Černý D, Near TJ.. 2018. Explosive diversification of marine fishes at the Cretaceous-Palaeogene boundary. Nat Ecol Evol. 2(4):688–696. - PubMed
-
- Alfaro ME, Holder MT.. 2006. The posterior and the prior in Bayesian phylogenetics. Annu Rev Ecol Evol Syst. 37(1):19–42.
-
- Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB.. 2002. Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst. 33(1):707–740.
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
