

Deciphering UV-induced DNA Damage Responses to Prevent and Treat Skin Cancer
- PMID: 32119110
- PMCID: PMC7651136
- DOI: 10.1111/php.13245
Deciphering UV-induced DNA Damage Responses to Prevent and Treat Skin Cancer
Abstract
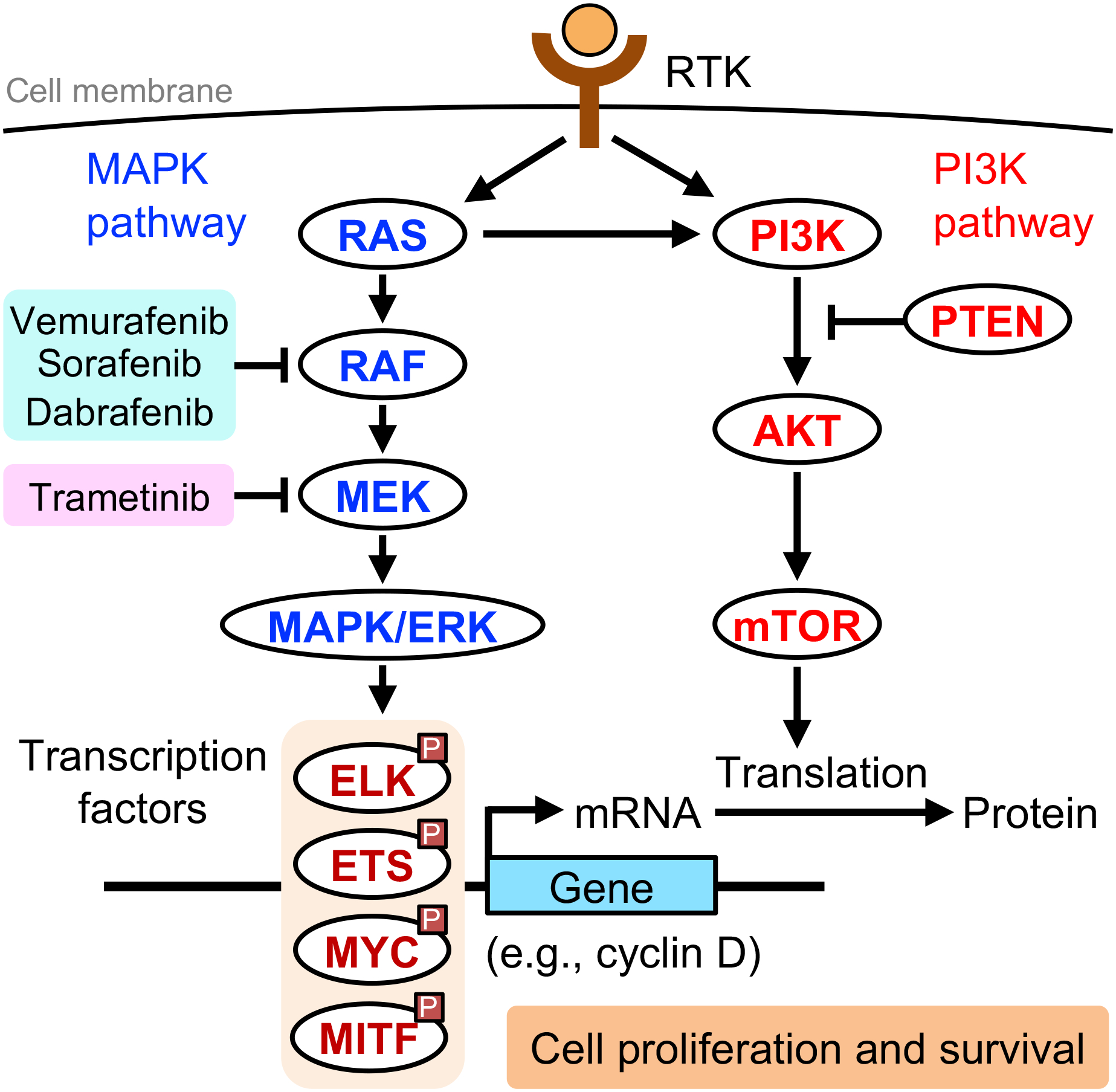
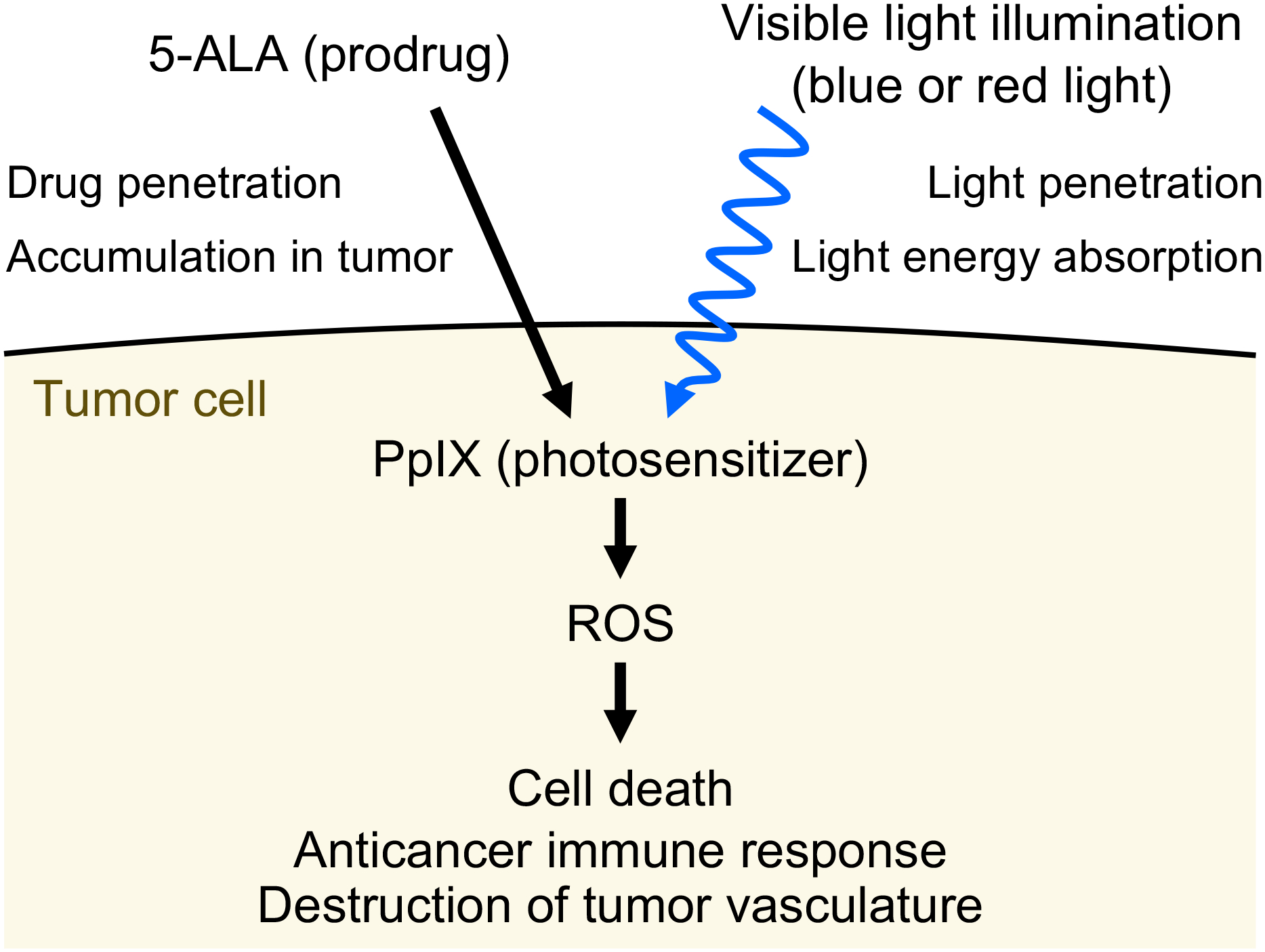
Ultraviolet (UV) radiation is among the most prevalent environmental factors that influence human health and disease. Even 1 h of UV irradiation extensively damages the genome. To cope with resulting deleterious DNA lesions, cells activate a multitude of DNA damage response pathways, including DNA repair. Strikingly, UV-induced DNA damage formation and repair are affected by chromatin state. When cells enter S phase with these lesions, a distinct mutation signature is created via error-prone translesion synthesis. Chronic UV exposure leads to high mutation burden in skin and consequently the development of skin cancer, the most common cancer in the United States. Intriguingly, UV-induced oxidative stress has opposing effects on carcinogenesis. Elucidating the molecular mechanisms of UV-induced DNA damage responses will be useful for preventing and treating skin cancer with greater precision. Excitingly, recent studies have uncovered substantial depth of novel findings regarding the molecular and cellular consequences of UV irradiation. In this review, we will discuss updated mechanisms of UV-induced DNA damage responses including the ATR pathway, which maintains genome integrity following UV irradiation. We will also present current strategies for preventing and treating nonmelanoma skin cancer, including ATR pathway inhibition for prevention and photodynamic therapy for treatment.
© 2020 American Society for Photobiology.
Conflict of interest statement
Competing Interests: The authors declare no competing interests.
Figures




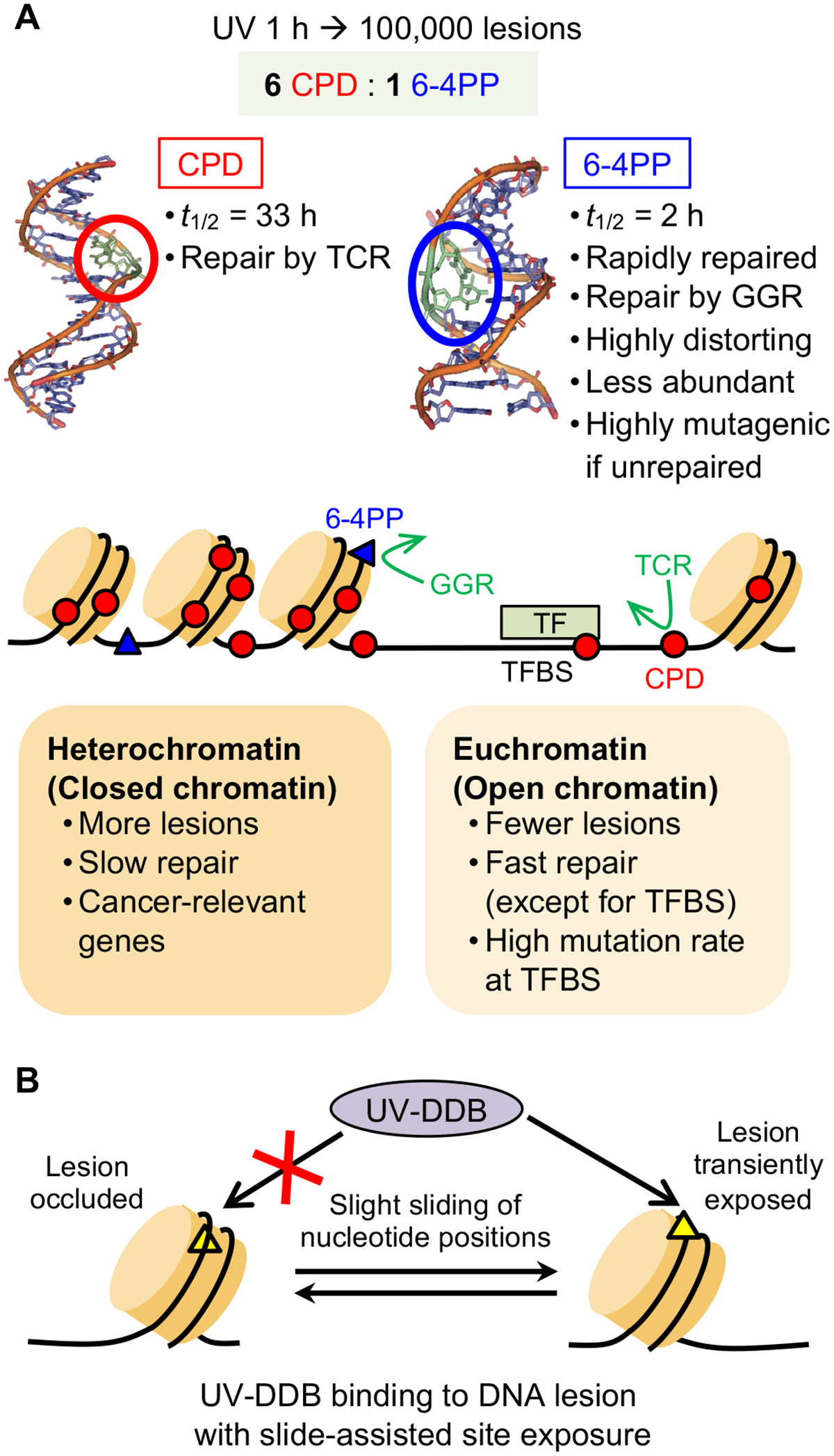
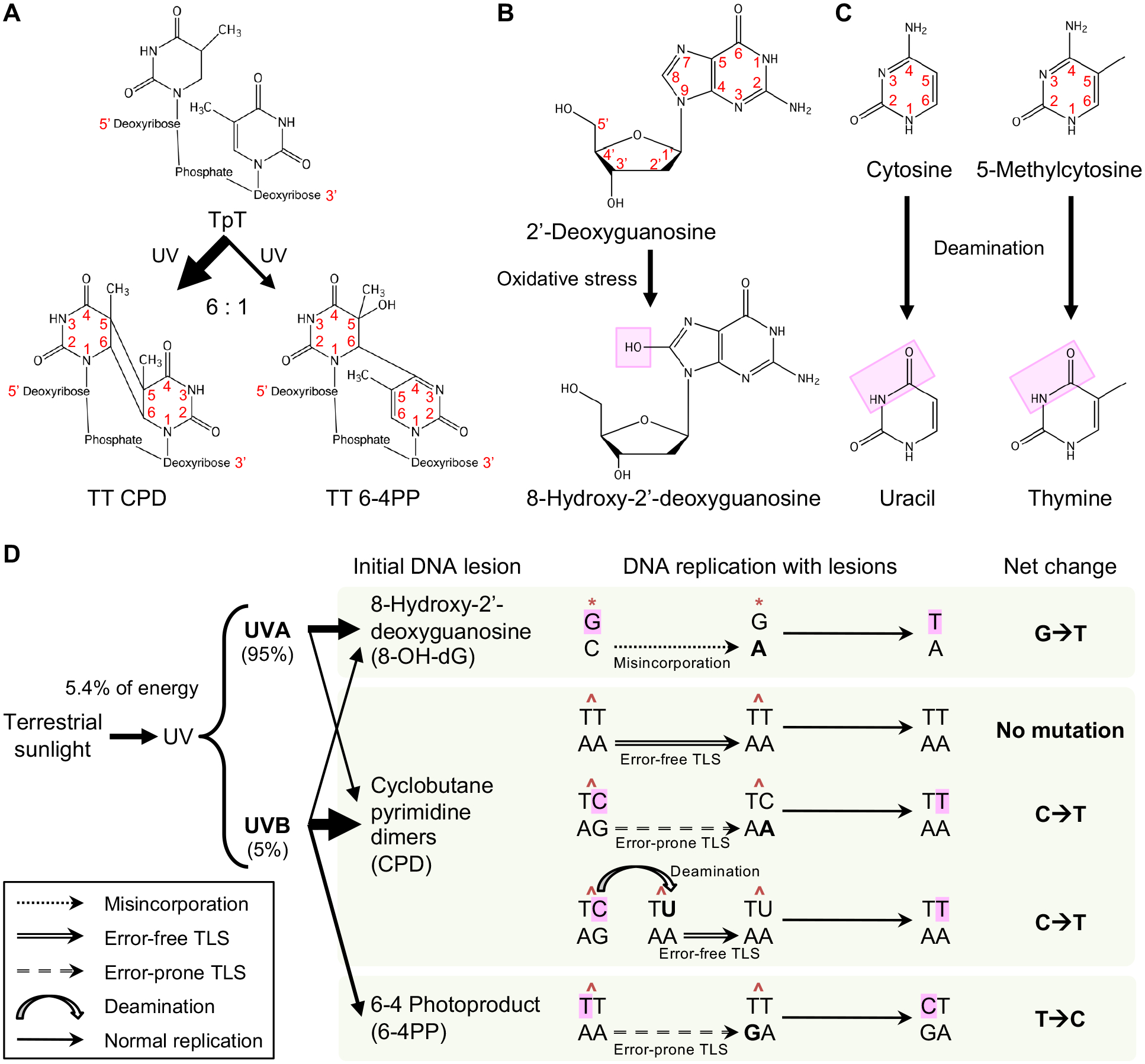
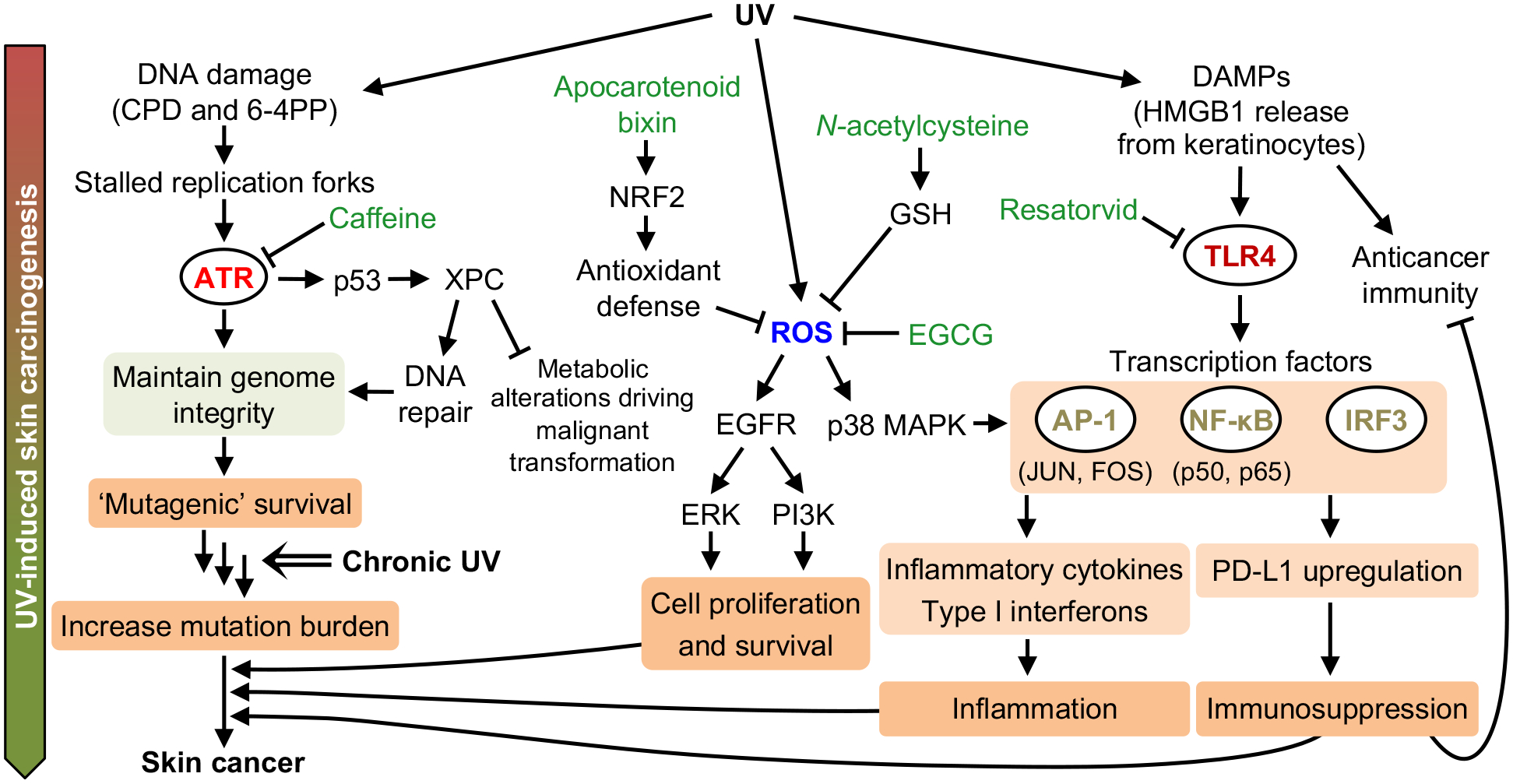
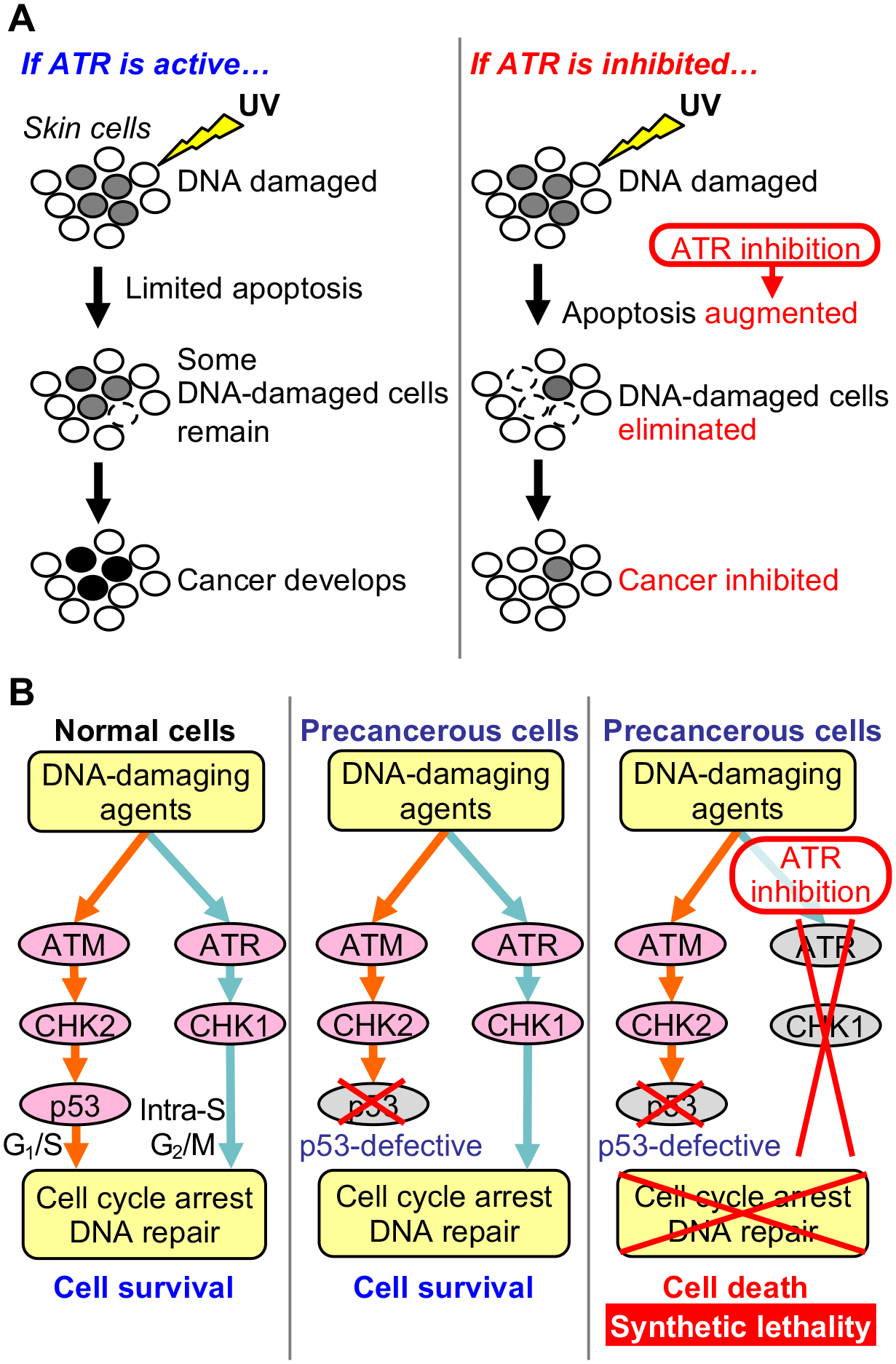
References
-
- Kwa RE, Campana K and Moy RL (1992) Biology of cutaneous squamous cell carcinoma. J Am Acad Dermatol 26, 1–26. - PubMed
-
- Albert MR and Ostheimer KG (2002) The evolution of current medical and popular attitudes toward ultraviolet light exposure: part 1. J Am Acad Dermatol 47, 930–937. - PubMed
-
- Armstrong BK, Kricker A and English DR (1997) Sun exposure and skin cancer. Australas J Dermatol 38 Suppl 1, S1–6. - PubMed
-
- Clydesdale GJ, Dandie GW and Muller HK (2001) Ultraviolet light induced injury: immunological and inflammatory effects. Immunol Cell Biol 79, 547–568. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
