

Machine learning with random subspace ensembles identifies antimicrobial resistance determinants from pan-genomes of three pathogens
- PMID: 32119670
- PMCID: PMC7067475
- DOI: 10.1371/journal.pcbi.1007608
Machine learning with random subspace ensembles identifies antimicrobial resistance determinants from pan-genomes of three pathogens
Abstract
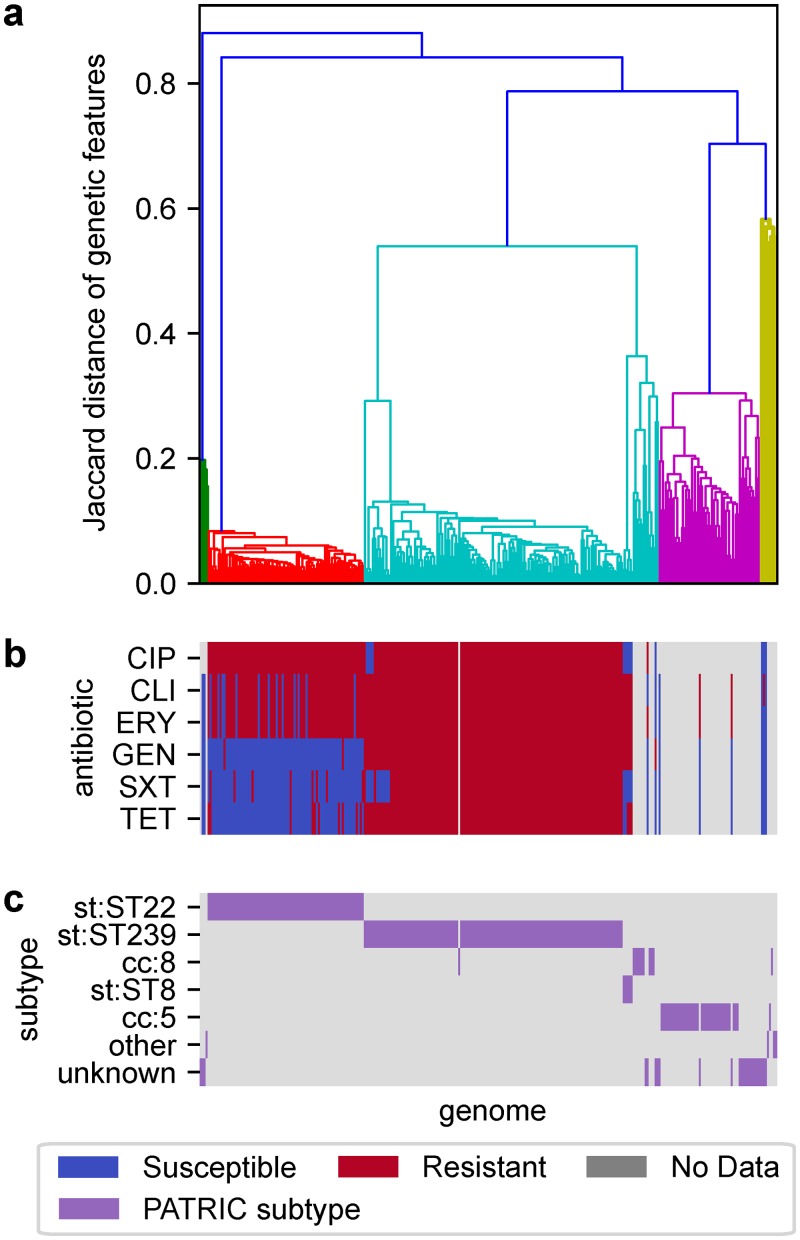
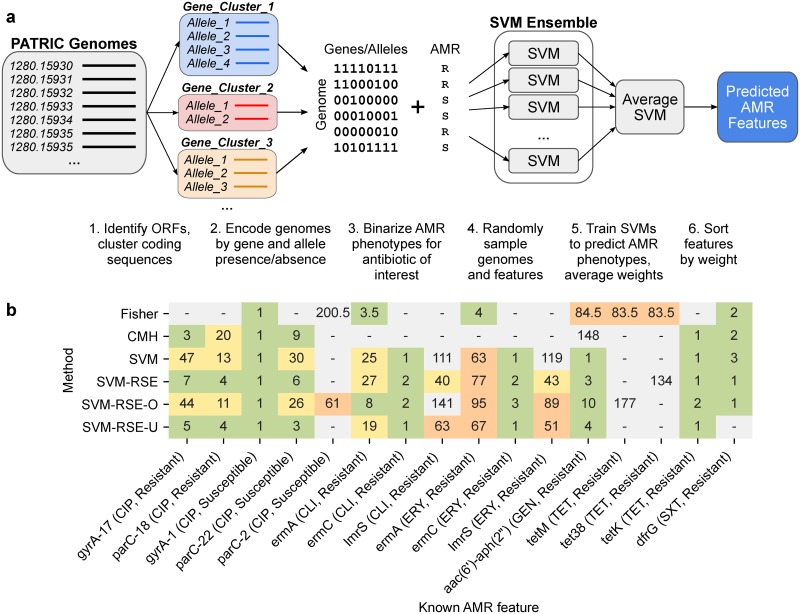
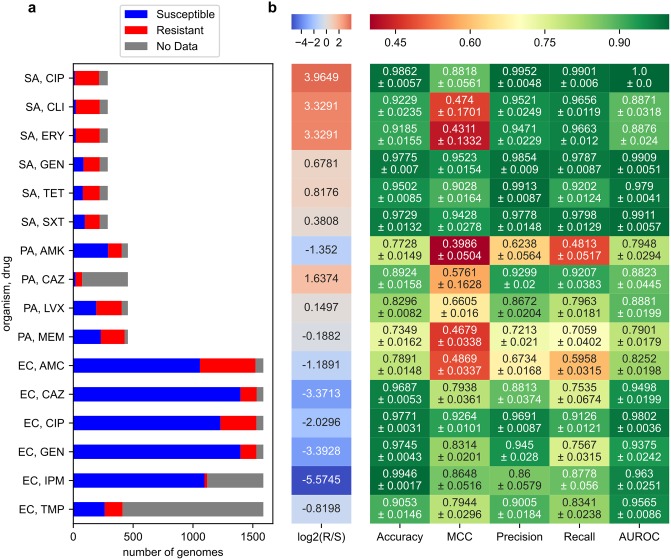
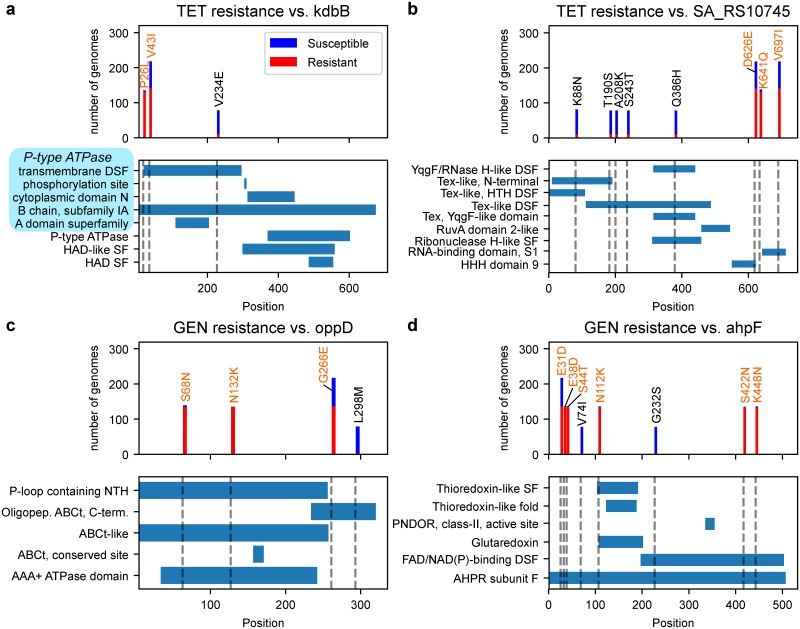
The evolution of antimicrobial resistance (AMR) poses a persistent threat to global public health. Sequencing efforts have already yielded genome sequences for thousands of resistant microbial isolates and require robust computational tools to systematically elucidate the genetic basis for AMR. Here, we present a generalizable machine learning workflow for identifying genetic features driving AMR based on constructing reference strain-agnostic pan-genomes and training random subspace ensembles (RSEs). This workflow was applied to the resistance profiles of 14 antimicrobials across three urgent threat pathogens encompassing 288 Staphylococcus aureus, 456 Pseudomonas aeruginosa, and 1588 Escherichia coli genomes. We find that feature selection by RSE detects known AMR associations more reliably than common statistical tests and previous ensemble approaches, identifying a total of 45 known AMR-conferring genes and alleles across the three organisms, as well as 25 candidate associations backed by domain-level annotations. Furthermore, we find that results from the RSE approach are consistent with existing understanding of fluoroquinolone (FQ) resistance due to mutations in the main drug targets, gyrA and parC, in all three organisms, and suggest the mutational landscape of those genes with respect to FQ resistance is simple. As larger datasets become available, we expect this approach to more reliably predict AMR determinants for a wider range of microbial pathogens.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
