

Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis
- PMID: 32121108
- PMCID: PMC7140469
- DOI: 10.3390/cells9030571
Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis
Abstract
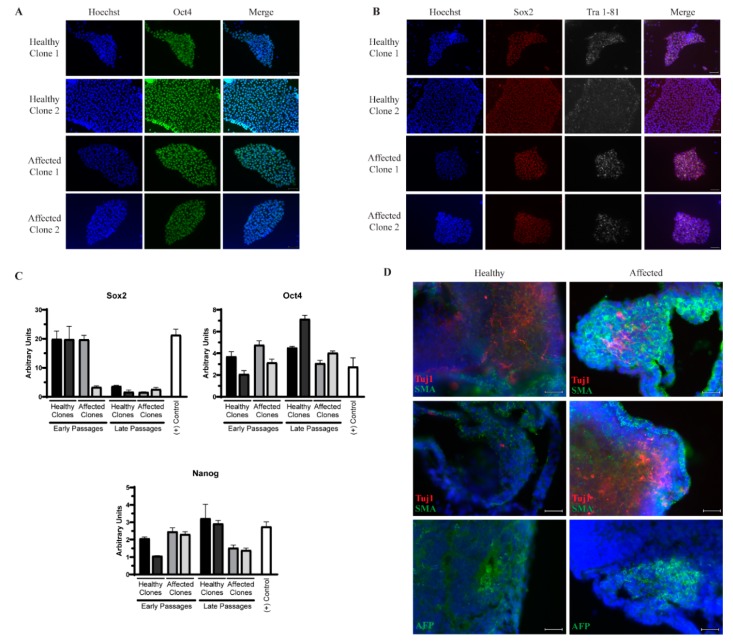
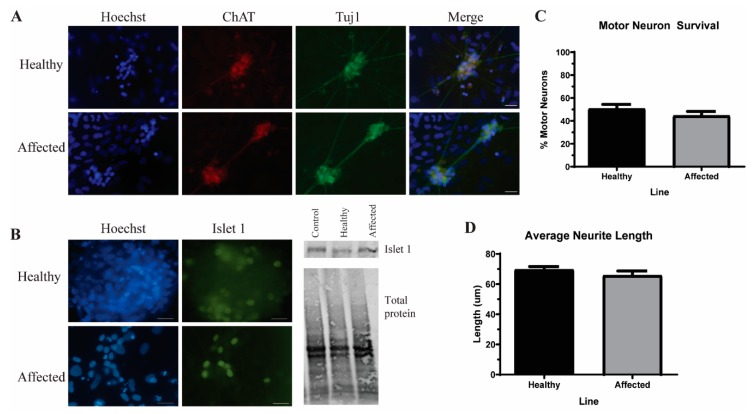
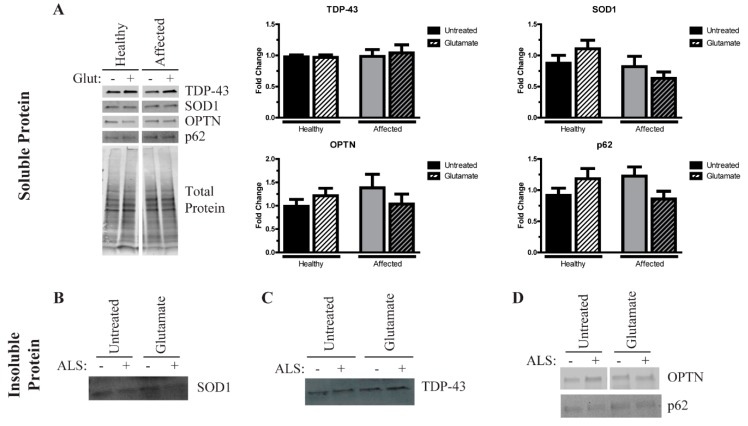
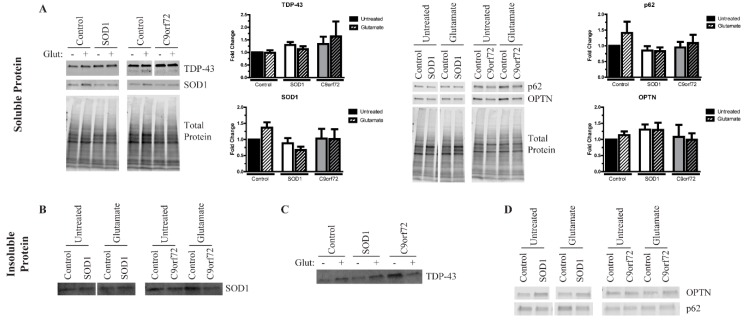
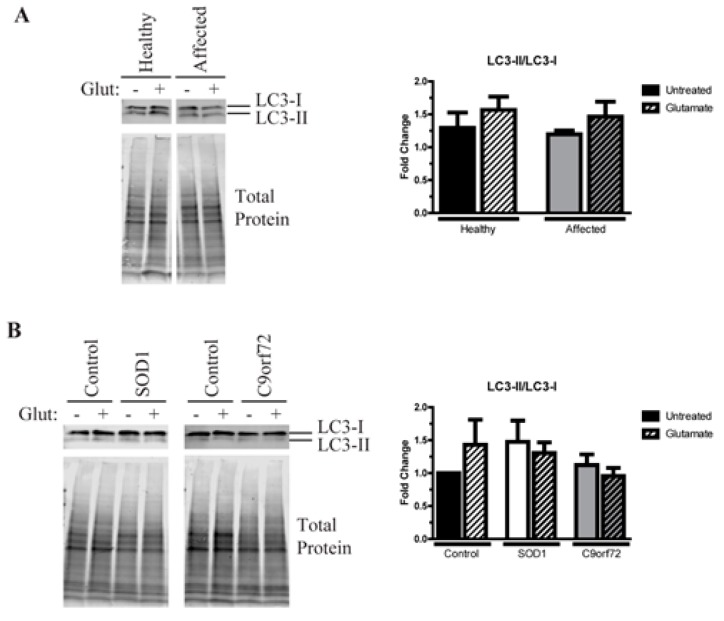
Amyotrophic lateral sclerosis (ALS) is a complex neurodegenerative disorder characterized by the loss of the upper and lower motor neurons. Approximately 10% of cases are caused by specific mutations in known genes, with the remaining cases having no known genetic link. As such, sporadic cases have been more difficult to model experimentally. Here, we describe the generation and differentiation of ALS induced pluripotent stem cells reprogrammed from discordant identical twins. Whole genome sequencing revealed no relevant mutations in known ALS-causing genes that differ between the twins. As protein aggregation is found in all ALS patients and is thought to contribute to motor neuron death, we sought to characterize the aggregation phenotype of the sporadic ALS induced pluripotent stem cells (iPSCs). Motor neurons from both twins had high levels of insoluble proteins that commonly aggregate in ALS that did not robustly change in response to exogenous glutamate. In contrast, established genetic ALS iPSC lines demonstrated insolubility in a protein- and genotype-dependent manner. Moreover, whereas the genetic ALS lines failed to induce autophagy after glutamate stress, motor neurons from both twins and independent controls did activate this protective pathway. Together, these data indicate that our unique model of sporadic ALS may provide key insights into disease pathology and highlight potential differences between sporadic and familial ALS.
Keywords: C9orf72; SOD1; familial ALS; glutamate toxicity; induced pluripotent stem cells; protein aggregation; sporadic ALS.
Conflict of interest statement
DF has received personal fees from serving on the Biogen ALS Advisory Board. The authors declare no other conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells.Mol Cell Neurosci. 2013 Sep;56:355-64. doi: 10.1016/j.mcn.2013.07.007. Epub 2013 Jul 25. Mol Cell Neurosci. 2013. PMID: 23891805 Free PMC article.
-
Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells.Stem Cell Res Ther. 2018 Nov 15;9(1):315. doi: 10.1186/s13287-018-1048-1. Stem Cell Res Ther. 2018. PMID: 30442180 Free PMC article.
-
Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis.Brain. 2020 Feb 1;143(2):430-440. doi: 10.1093/brain/awz419. Brain. 2020. PMID: 32040555 Free PMC article.
-
ALS, a cellular whodunit on motor neuron degeneration.Mol Cell Neurosci. 2020 Sep;107:103524. doi: 10.1016/j.mcn.2020.103524. Epub 2020 Jul 3. Mol Cell Neurosci. 2020. PMID: 32629110 Review.
-
Reverse engineering human neurodegenerative disease using pluripotent stem cell technology.Brain Res. 2016 May 1;1638(Pt A):30-41. doi: 10.1016/j.brainres.2015.09.023. Epub 2015 Sep 28. Brain Res. 2016. PMID: 26423934 Free PMC article. Review.
Cited by
-
ACTA1 H40Y mutant iPSC-derived skeletal myocytes display mitochondrial defects in an in vitro model of nemaline myopathy.Exp Cell Res. 2023 Mar 15;424(2):113507. doi: 10.1016/j.yexcr.2023.113507. Epub 2023 Feb 14. Exp Cell Res. 2023. PMID: 36796746 Free PMC article.
-
Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons.Genes (Basel). 2022 Jan 27;13(2):241. doi: 10.3390/genes13020241. Genes (Basel). 2022. PMID: 35205286 Free PMC article.
-
Stem Cell-Based Disease Modeling and Cell Therapy.Cells. 2020 Sep 29;9(10):2193. doi: 10.3390/cells9102193. Cells. 2020. PMID: 33003295 Free PMC article.
References
-
- Mackenzie I.R., Bigio E.H., Ince P.G., Geser F., Neumann M., Cairns N.J., Kwong L.K., Forman M.S., Ravits J., Stewart H., et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. - DOI - PubMed
-
- Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
