

Integrating Family-Based and Mendelian Randomization Designs
- PMID: 32122917
- PMCID: PMC7919398
- DOI: 10.1101/cshperspect.a039503
Integrating Family-Based and Mendelian Randomization Designs
Abstract
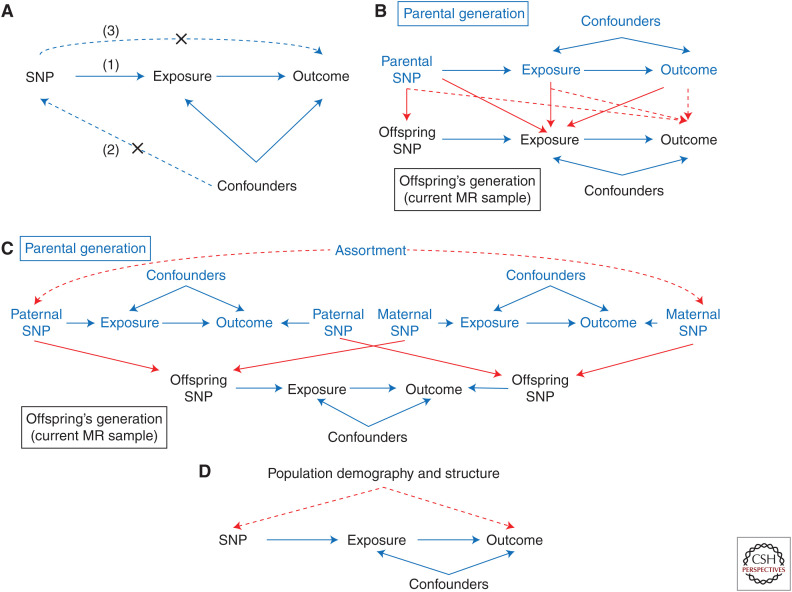
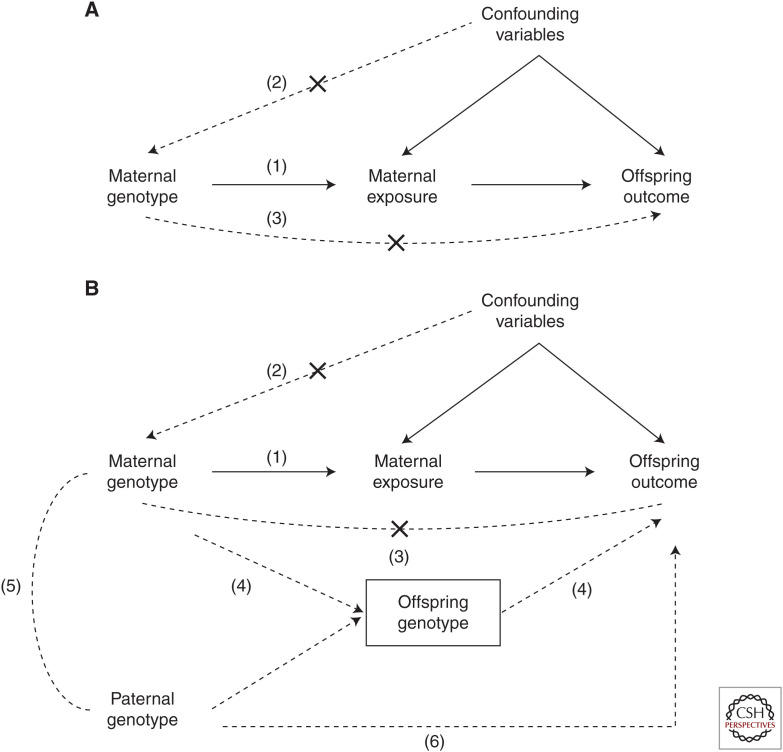
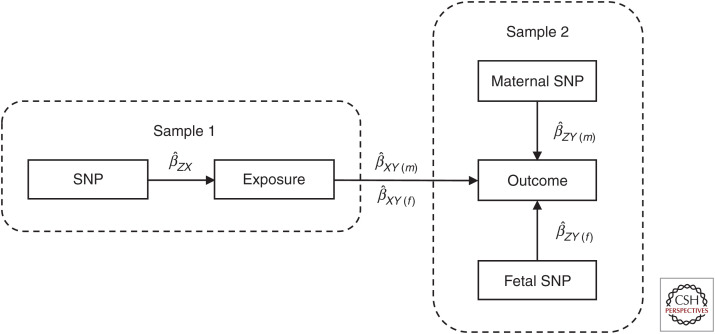
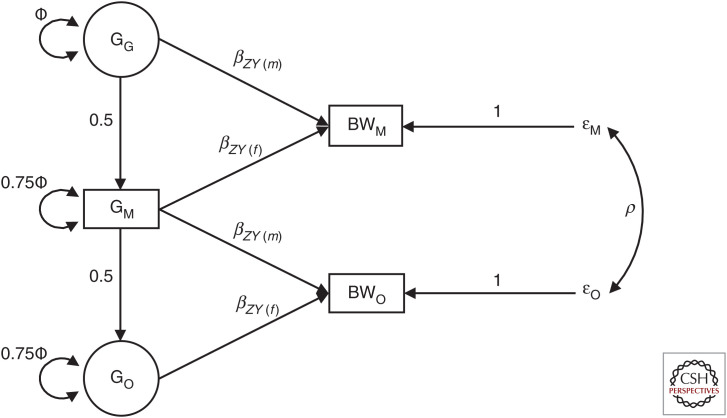
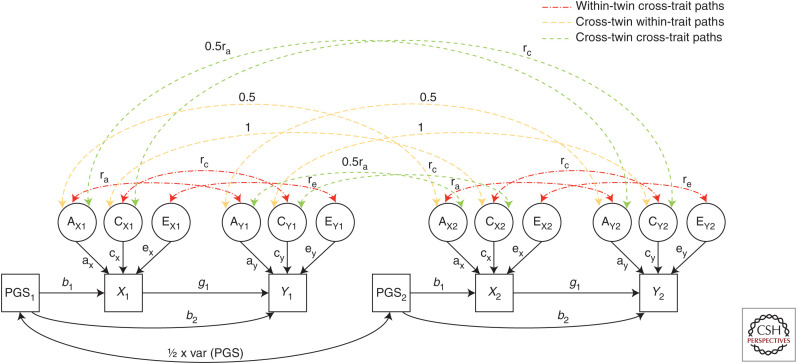
Most Mendelian randomization (MR) studies published in the literature to date have involved analyses of unrelated, putatively independent sets of individuals. However, estimates obtained from these sorts of studies are subject to a range of biases including dynastic effects, assortative mating, residual population stratification, and horizontal pleiotropy. The inclusion of related individuals in MR studies can help control for and, in some cases, estimate the effect of these biases on causal parameters. In this review, we discuss these biases, how they can affect MR studies, and describe three sorts of family-based study designs that can be used to control for them. We conclude that including family information from related individuals is not only possible given the world's existing twin, birth, and large-scale population-based cohorts, but likely to reap rich rewards in understanding the etiology of complex traits and diseases in the near future.
Copyright © 2021 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Bates TC, Maher BS, Medland SE, McAloney K, Wright MJ, Hansell NK, Kendler KS, Martin NG, Gillespie NA. 2018. The nature of nurture: using a virtual-parent design to test parenting effects on children's educational attainment in genotyped families. Twin Res Hum Genet 21: 73–83. 10.1017/thg.2018.11 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical