

Integrated structural and evolutionary analysis reveals common mechanisms underlying adaptive evolution in mammals
- PMID: 32123117
- PMCID: PMC7084095
- DOI: 10.1073/pnas.1916786117
Integrated structural and evolutionary analysis reveals common mechanisms underlying adaptive evolution in mammals
Abstract
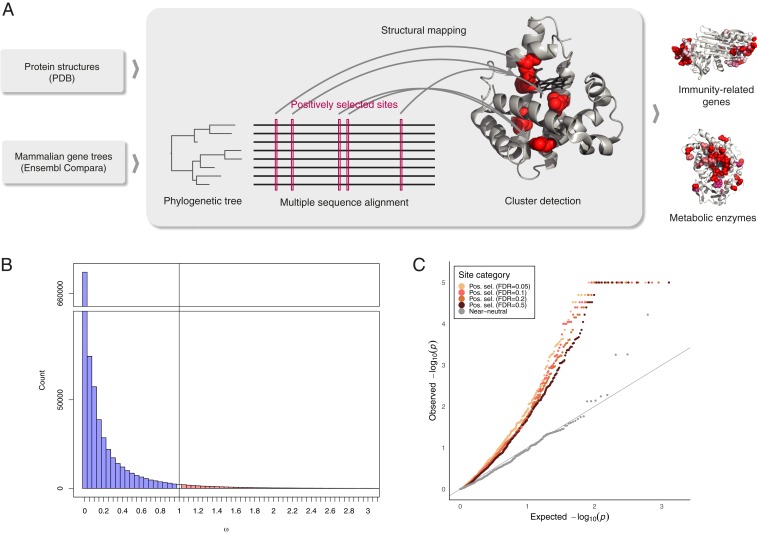
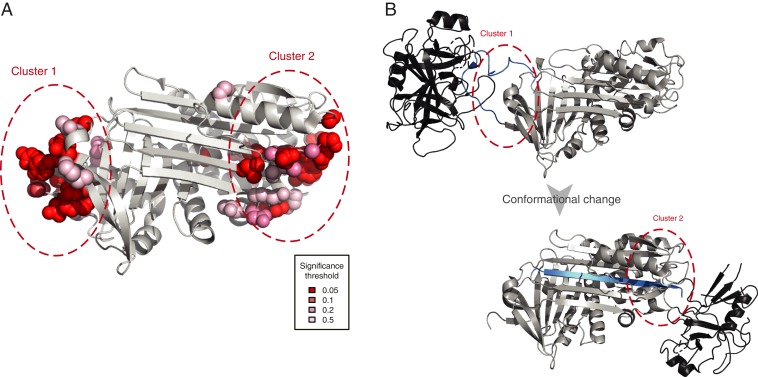
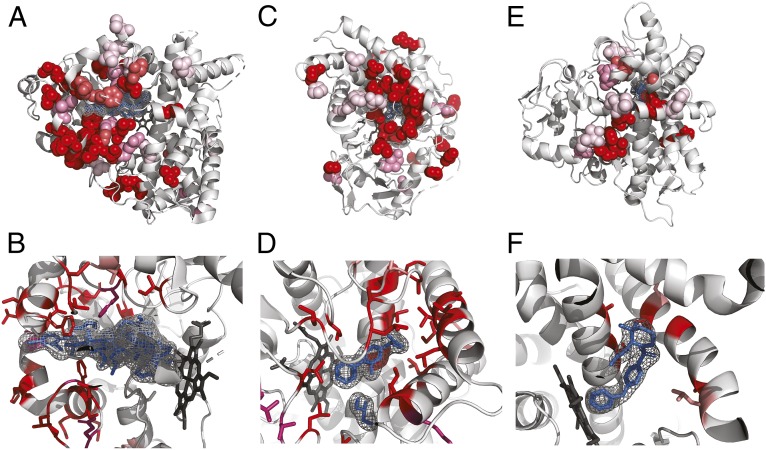
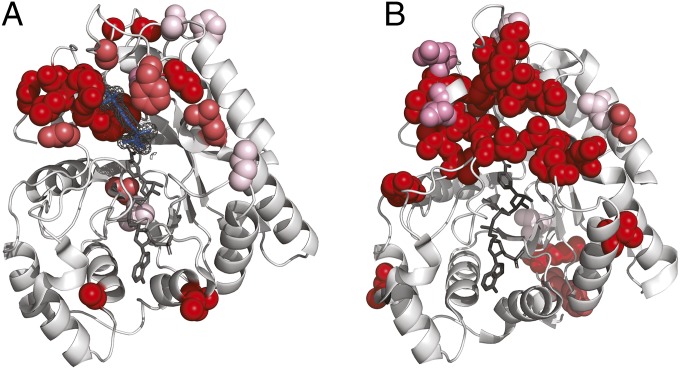
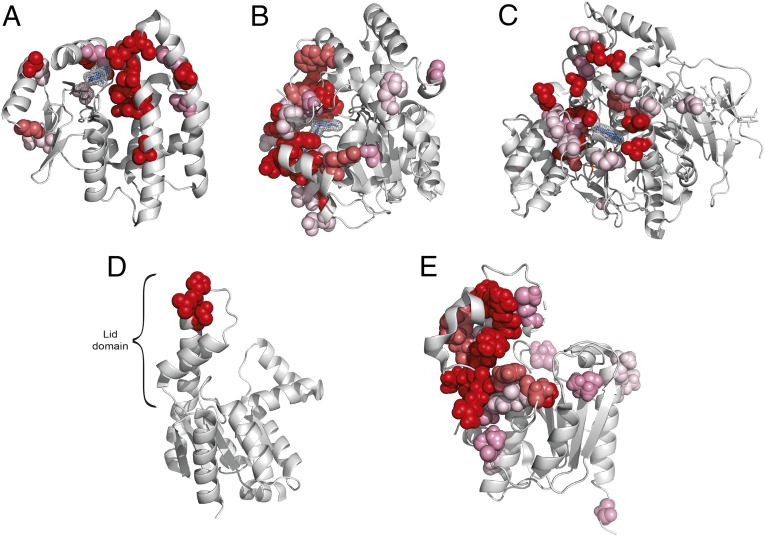
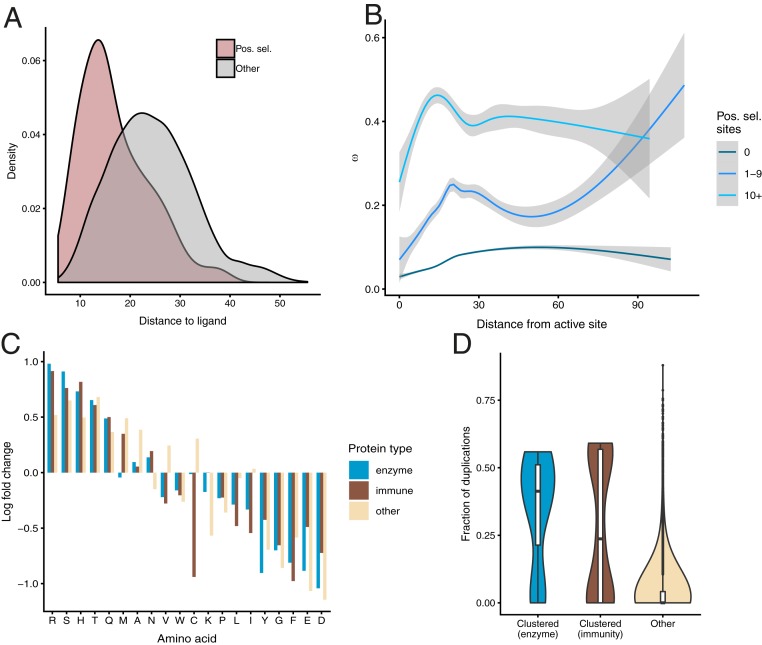
Understanding the molecular basis of adaptation to the environment is a central question in evolutionary biology, yet linking detected signatures of positive selection to molecular mechanisms remains challenging. Here we demonstrate that combining sequence-based phylogenetic methods with structural information assists in making such mechanistic interpretations on a genomic scale. Our integrative analysis shows that positively selected sites tend to colocalize on protein structures and that positively selected clusters are found in functionally important regions of proteins, indicating that positive selection can contravene the well-known principle of evolutionary conservation of functionally important regions. This unexpected finding, along with our discovery that positive selection acts on structural clusters, opens previously unexplored strategies for the development of better models of protein evolution. Remarkably, proteins where we detect the strongest evidence of clustering belong to just two functional groups: Components of immune response and metabolic enzymes. This gives a coherent picture of pathogens and xenobiotics as important drivers of adaptive evolution of mammals.
Keywords: adaptive evolution; immunity; mammals; metabolism; protein evolution.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
