

The structure of microbial populations in Nelore GIT reveals inter-dependency of methanogens in feces and rumen
- PMID: 32123563
- PMCID: PMC7038601
- DOI: 10.1186/s40104-019-0422-x
The structure of microbial populations in Nelore GIT reveals inter-dependency of methanogens in feces and rumen
Abstract
Background: The success of different species of ruminants in the colonization of a diverse range of environments is due to their ability to digest and absorb nutrients from cellulose, a complex polysaccharide found in leaves and grass. Ruminants rely on a complex and diverse microbial community, or microbiota, in a unique compartment known as the rumen to break down this polysaccharide. Changes in microbial populations of the rumen can affect the host's development, health, and productivity. However, accessing the rumen is stressful for the animal. Therefore, the development and use of alternative sampling methods are needed if this technique is to be routinely used in cattle breeding. To this end, we tested if the fecal microbiome could be used as a proxy for the rumen microbiome due to its accessibility. We investigated the taxonomic composition, diversity and inter-relations of two different GIT compartments, rumen and feces, of 26 Nelore (Bos indicus) bulls, using Next Generation Sequencing (NGS) metabarcoding of bacteria, archaea and ciliate protozoa.
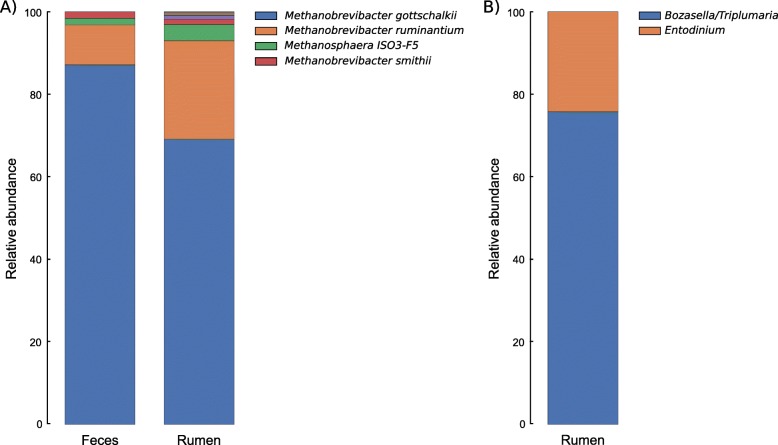
Results: We identified 4265 Amplicon Sequence Variants (ASVs) from bacteria, 571 from archaea, and 107 from protozoa, of which 143 (96 bacteria and 47 archaea) were found common between both microbiomes. The most prominent bacterial phyla identified were Bacteroidetes (41.48%) and Firmicutes (56.86%) in the ruminal and fecal microbiomes, respectively, with Prevotella and Ruminococcaceae UCG-005 the most relatively abundant genera identified in each microbiome. The most abundant archaeal phylum identified was Euryarchaeota, of which Methanobrevibacter gottschalkii, a methanogen, was the prevalent archaeal species identified in both microbiomes. Protozoa were found exclusively identified in the rumen with Bozasella/Triplumaria being the most frequent genus identified. Co-occurrence among ruminal and fecal ASVs reinforces the relationship of microorganisms within a biological niche. Furthermore, the co-occurrence of shared archaeal ASVs between microbiomes indicates a dependency of the predominant fecal methanogen population on the rumen population.
Conclusions: Co-occurring microorganisms were identified within the rumen and fecal microbiomes, which revealed a strong association and inter-dependency between bacterial, archaeal and protozoan populations of the same microbiome. The archaeal ASVs identified as co-occurring between GIT compartments corresponded to the methanogenic genera Methanobrevibacter and Methanosphaera and represented 26.34% of the overall archaeal sequencesdiversity in the rumen and 42.73% in feces. Considering that these archaeal ASVs corresponded to a significant part of the overall diversity of both microbiomes, which is much higher if one includes the interactions of these co-occurring with other rumen archaea ASVs, we suggest that fecal methanogens could be used as a proxy of ruminal methanogens.
Keywords: Archaea; Bacteria; Bos indicus; Metabarcoding; Methanobrevibacter; Microbiome; Microbiota.
© The Author(s). 2020.
Conflict of interest statement
Competing interestsThe authors Paul Walsh and Polyana C. Tizioto were employed by the companies Nsilico and NGS Genomic Solutions, respectively. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


References
LinkOut - more resources
Full Text Sources
