

Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism
- PMID: 32123736
- PMCID: PMC7048371
- DOI: 10.1021/acscentsci.9b01071
Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism
Abstract
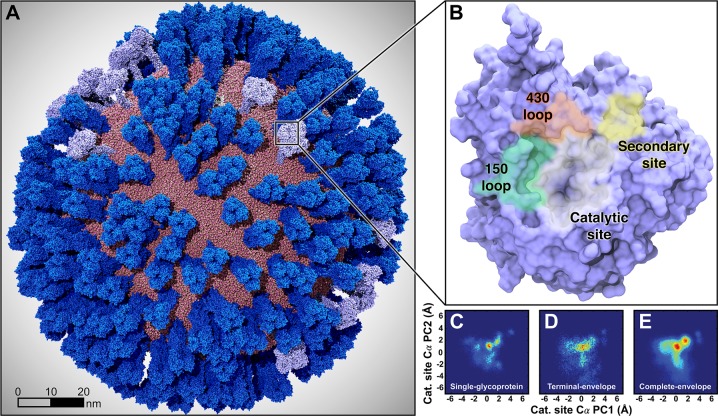
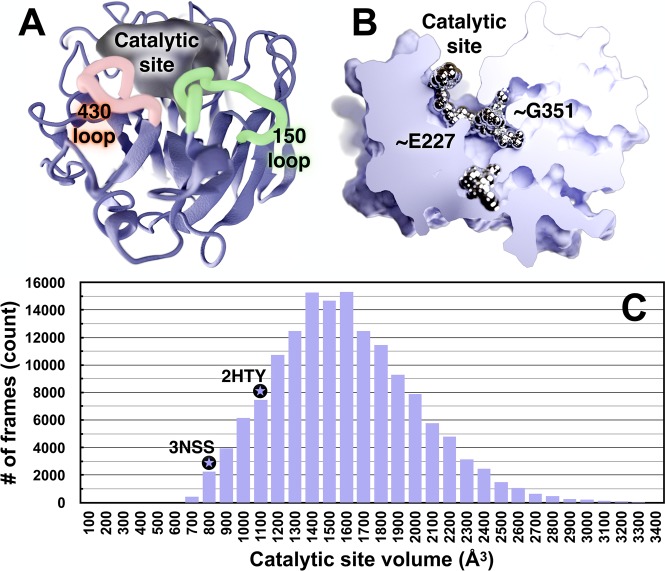
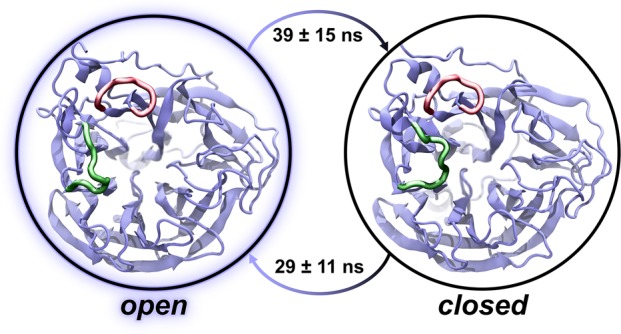
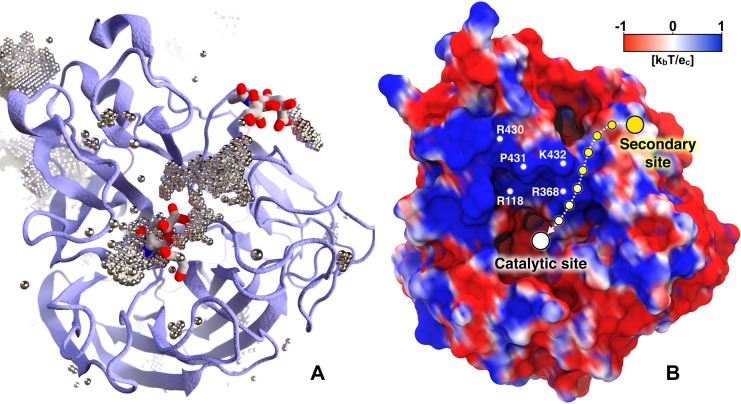
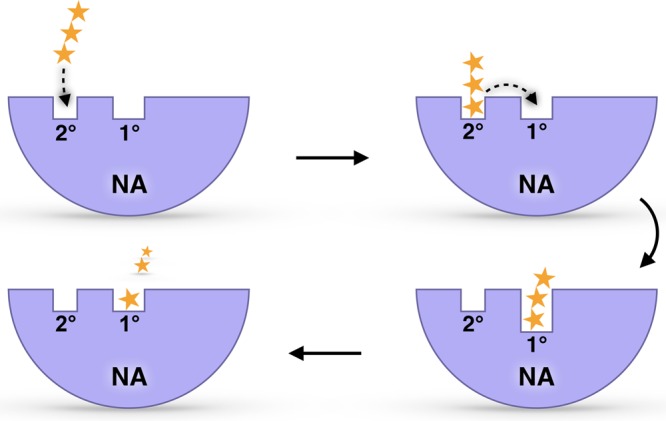
Influenza virus circulates in human, avian, and swine hosts, causing seasonal epidemic and occasional pandemic outbreaks. Influenza neuraminidase, a viral surface glycoprotein, has two sialic acid binding sites. The catalytic (primary) site, which also binds inhibitors such as oseltamivir carboxylate, is responsible for cleaving the sialic acid linkages that bind viral progeny to the host cell. In contrast, the functional annotation of the secondary site remains unclear. Here, we better characterize these two sites through the development of an all-atom, explicitly solvated, and experimentally based integrative model of the pandemic influenza A H1N1 2009 viral envelope, containing ∼160 million atoms and spanning ∼115 nm in diameter. Molecular dynamics simulations of this crowded subcellular environment, coupled with Markov state model theory, provide a novel framework for studying realistic molecular systems at the mesoscale and allow us to quantify the kinetics of the neuraminidase 150-loop transition between the open and closed states. An analysis of chloride ion occupancy along the neuraminidase surface implies a potential new role for the neuraminidase secondary site, wherein the terminal sialic acid residues of the linkages may bind before transfer to the primary site where enzymatic cleavage occurs. Altogether, our work breaks new ground for molecular simulation in terms of size, complexity, and methodological analyses of the components. It also provides fundamental insights into the understanding of substrate recognition processes for this vital influenza drug target, suggesting a new strategy for the development of anti-influenza therapeutics.
Copyright © 2020 American Chemical Society.
Conflict of interest statement
The authors declare the following competing financial interest(s): R.E.A. is a cofounder and on the scientific advisory board of, and has equity interest in, Actavalon, Inc.
Figures
References
-
- Centers for Disease Control and Prevention . Summary of the 2017–2018 Influenza Season; United States Centers for Disease Control and Prevention: 2018; Vol. 11, pp. 7–10.
