

Atm deficiency in the DNA polymerase β null cerebellum results in cerebellar ataxia and Itpr1 reduction associated with alteration of cytosine methylation
- PMID: 32123907
- PMCID: PMC7144915
- DOI: 10.1093/nar/gkaa140
Atm deficiency in the DNA polymerase β null cerebellum results in cerebellar ataxia and Itpr1 reduction associated with alteration of cytosine methylation
Abstract
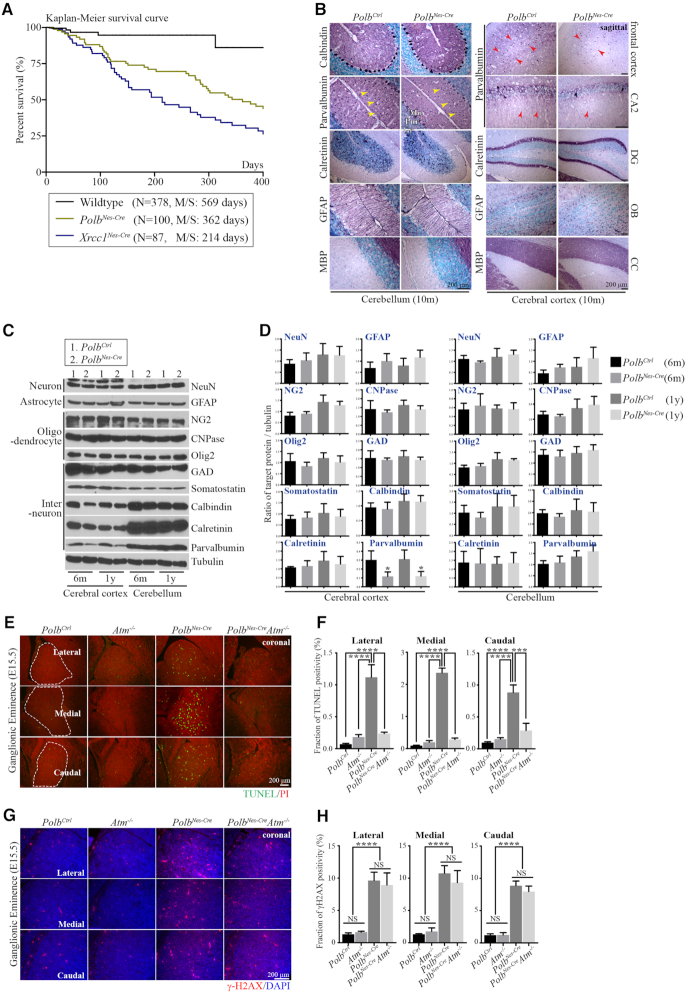
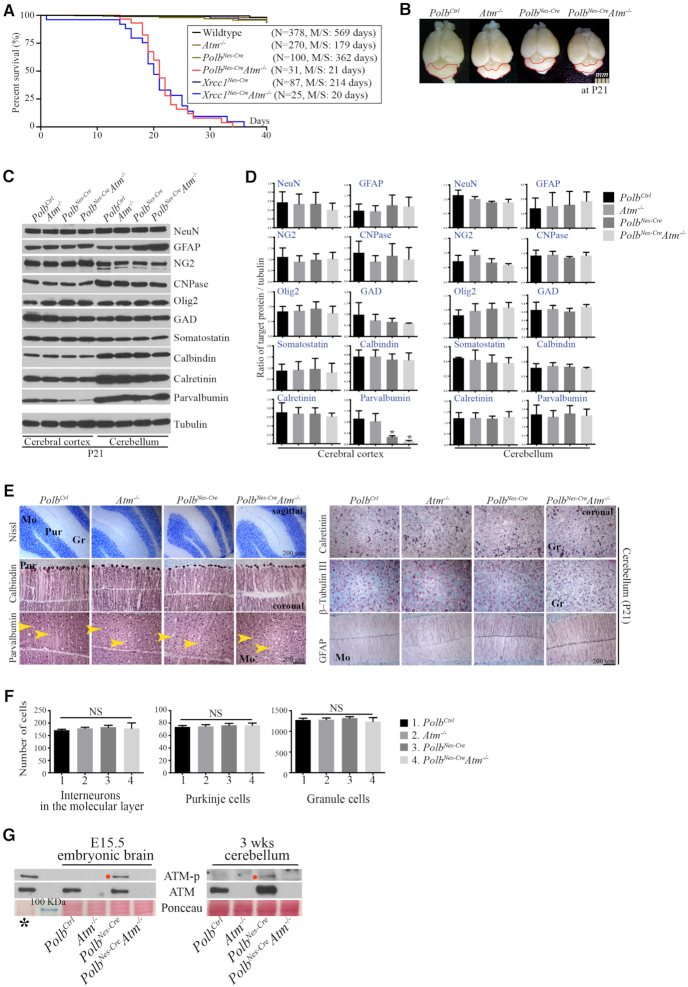
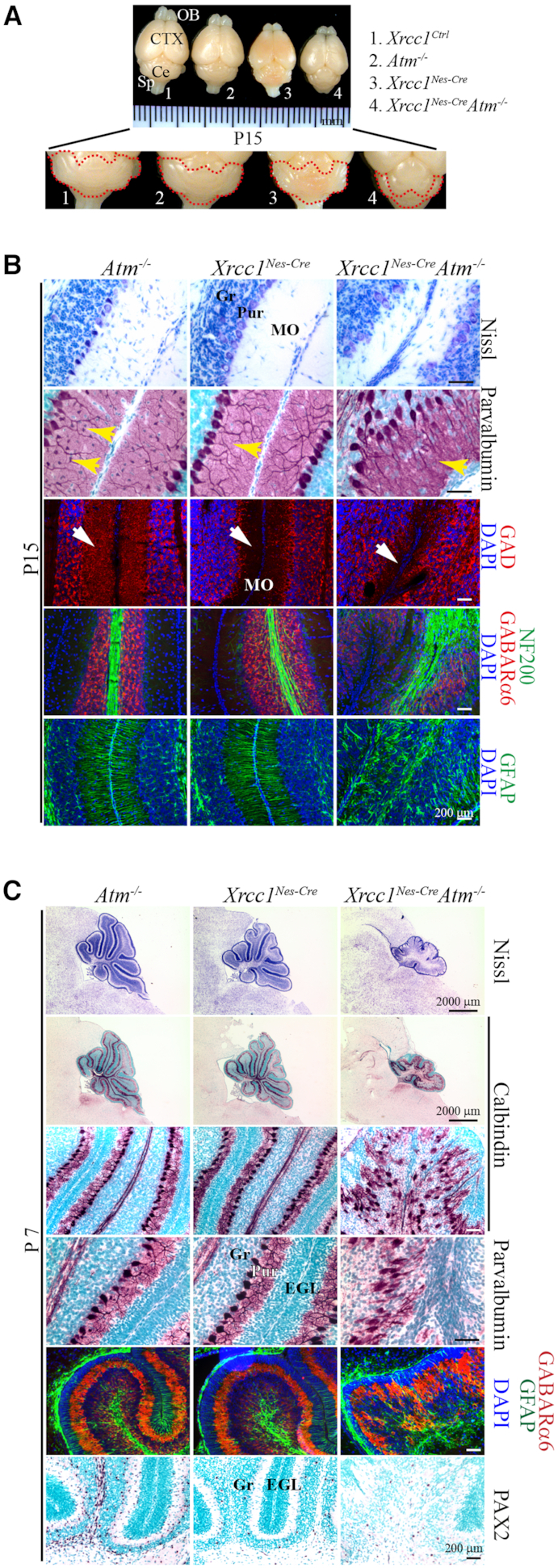
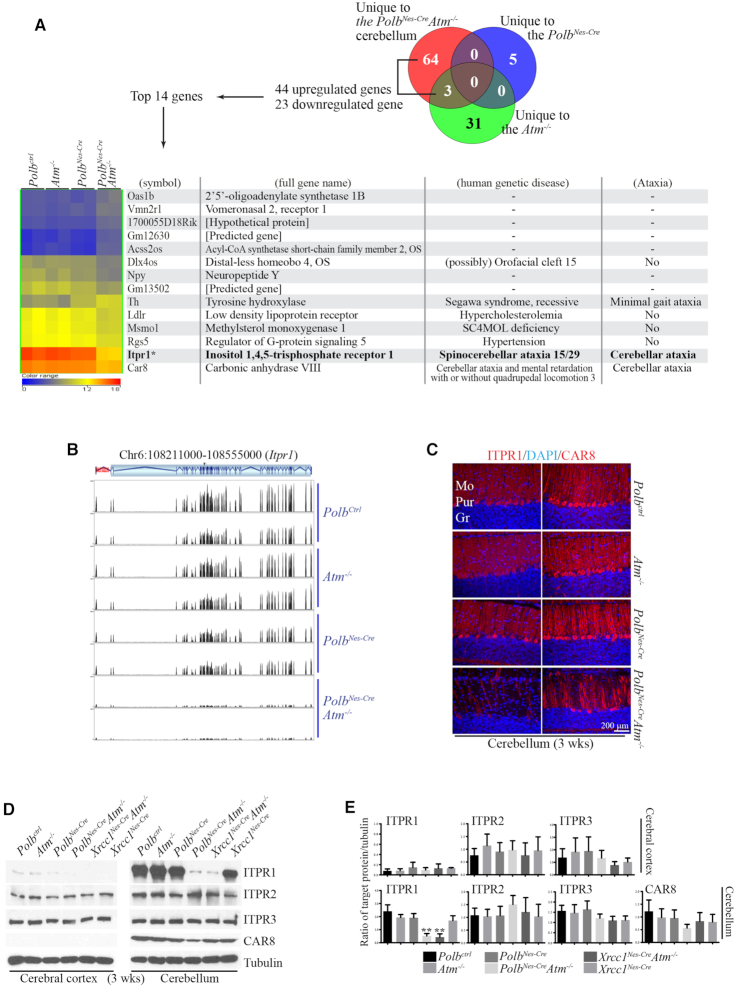
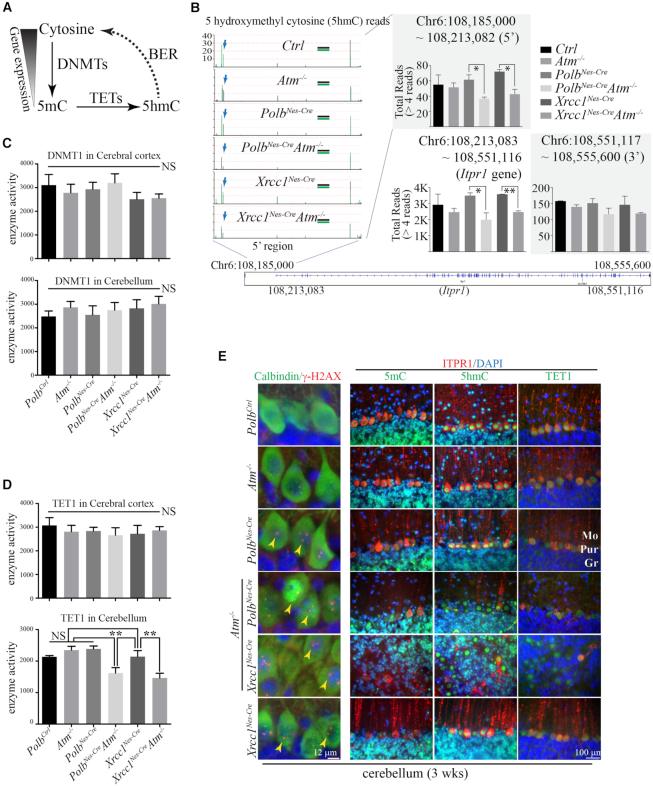
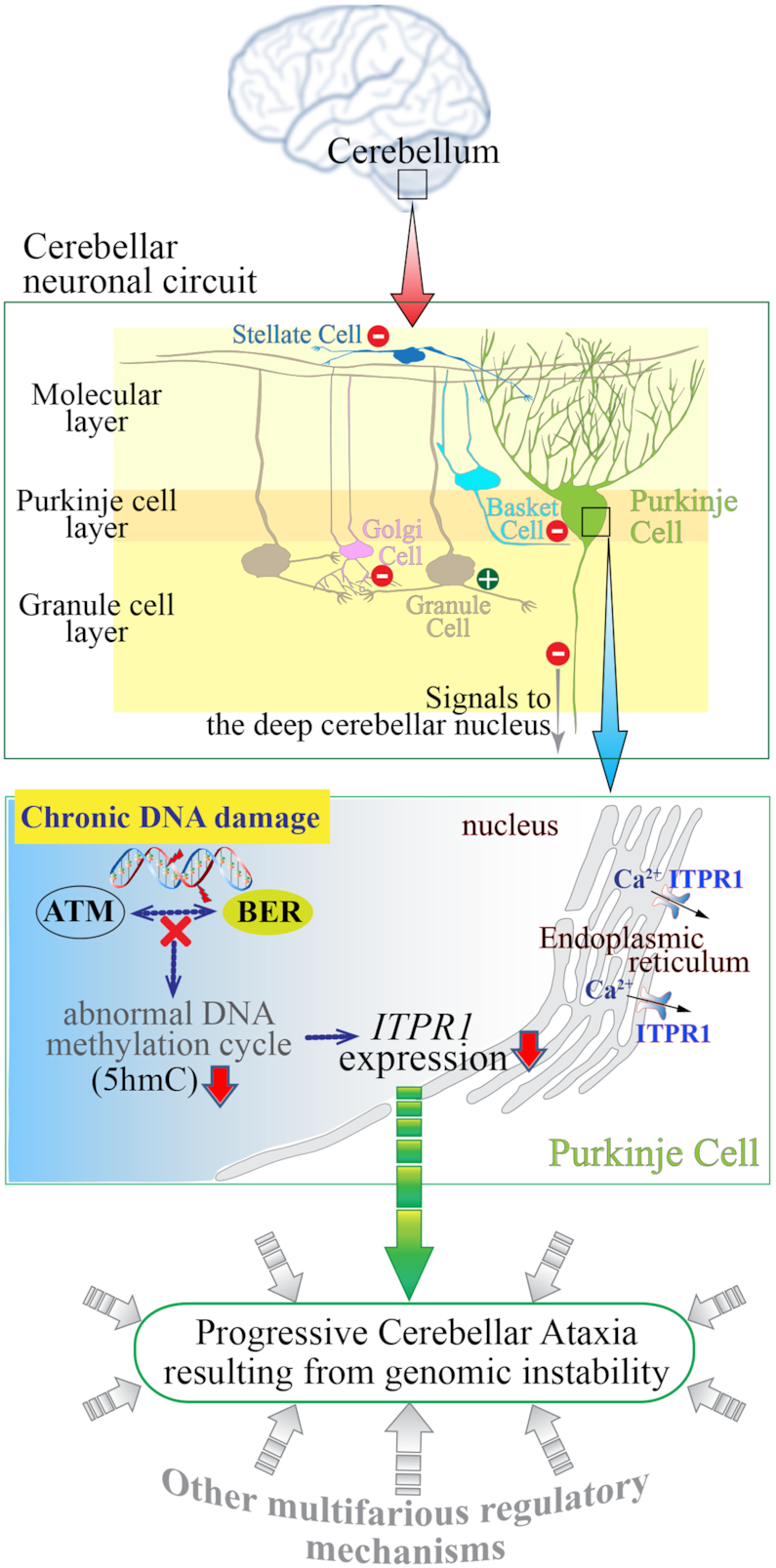
Genomic instability resulting from defective DNA damage responses or repair causes several abnormalities, including progressive cerebellar ataxia, for which the molecular mechanisms are not well understood. Here, we report a new murine model of cerebellar ataxia resulting from concomitant inactivation of POLB and ATM. POLB is one of key enzymes for the repair of damaged or chemically modified bases, including methylated cytosine, but selective inactivation of Polb during neurogenesis affects only a subpopulation of cortical interneurons despite the accumulation of DNA damage throughout the brain. However, dual inactivation of Polb and Atm resulted in ataxia without significant neuropathological defects in the cerebellum. ATM is a protein kinase that responds to DNA strand breaks, and mutations in ATM are responsible for Ataxia Telangiectasia, which is characterized by progressive cerebellar ataxia. In the cerebella of mice deficient for both Polb and Atm, the most downregulated gene was Itpr1, likely because of misregulated DNA methylation cycle. ITPR1 is known to mediate calcium homeostasis, and ITPR1 mutations result in genetic diseases with cerebellar ataxia. Our data suggest that dysregulation of ITPR1 in the cerebellum could be one of contributing factors to progressive ataxia observed in human genomic instability syndromes.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Lee Y., Choi I., Kim J., Kim K.. DNA damage to human genetic disorders with neurodevelopmental defects. J. Genet. Med. 2016; 13:1–13.
-
- Blackford A.N., Jackson S.P.. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell. 2017; 66:801–817. - PubMed
-
- McKinnon P.J., Caldecott K.W.. DNA strand break repair and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2007; 8:37–55. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
