

IRF4 instructs effector Treg differentiation and immune suppression in human cancer
- PMID: 32125291
- PMCID: PMC7260038
- DOI: 10.1172/JCI130426
IRF4 instructs effector Treg differentiation and immune suppression in human cancer
Abstract
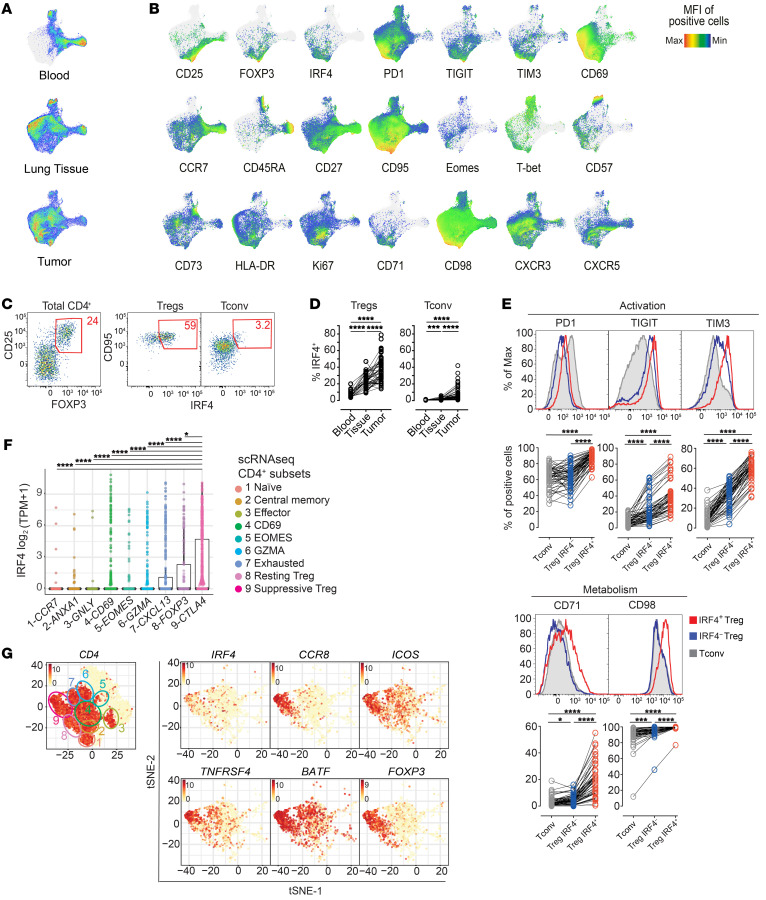
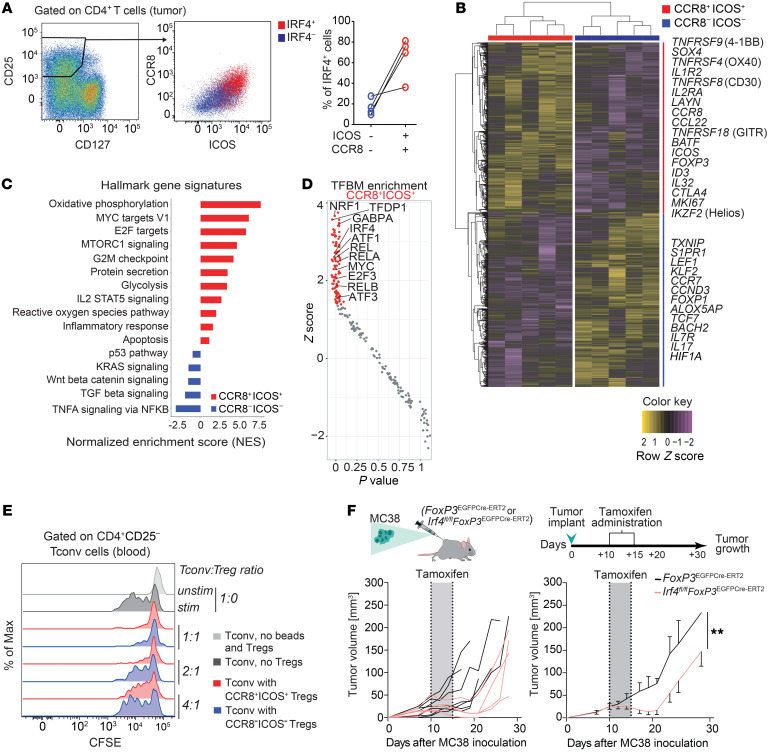
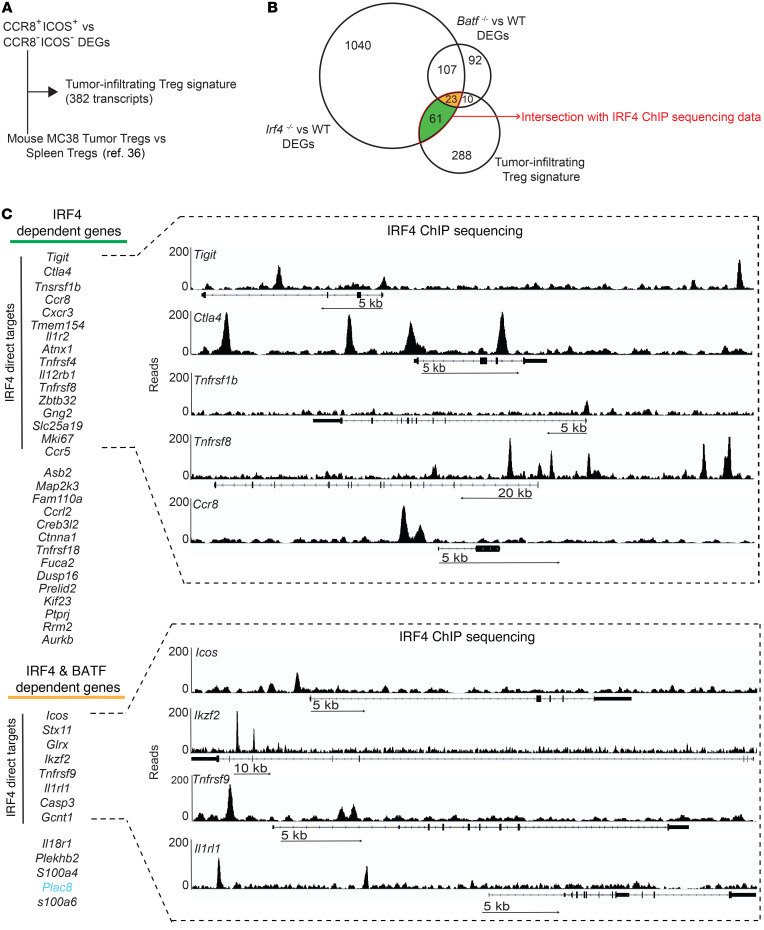
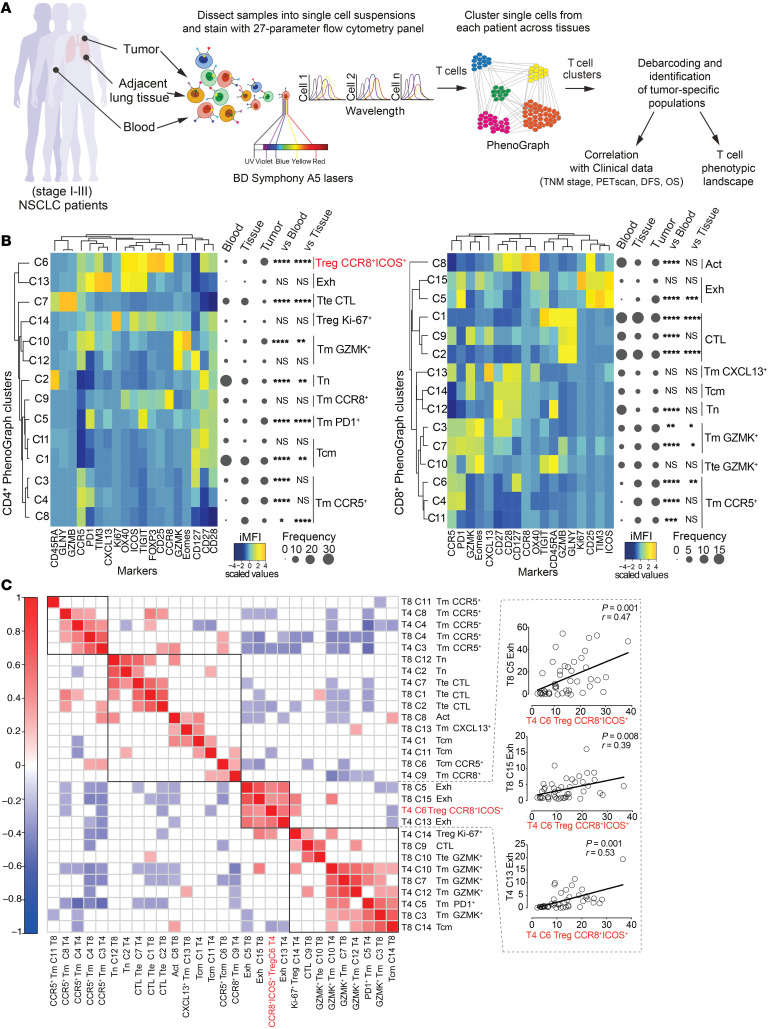
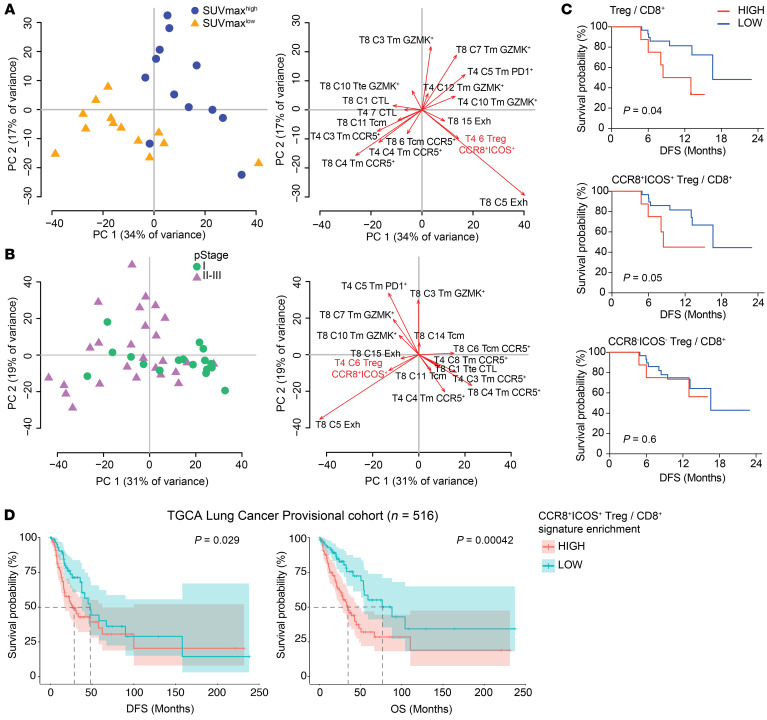
The molecular mechanisms responsible for the high immunosuppressive capacity of CD4+ Tregs in tumors are not well known. High-dimensional single-cell profiling of T cells from chemotherapy-naive individuals with non-small-cell lung cancer identified the transcription factor IRF4 as specifically expressed by a subset of intratumoral CD4+ effector Tregs with superior suppressive activity. In contrast to the IRF4- counterparts, IRF4+ Tregs expressed a vast array of suppressive molecules, and their presence correlated with multiple exhausted subpopulations of T cells. Integration of transcriptomic and epigenomic data revealed that IRF4, either alone or in combination with its partner BATF, directly controlled a molecular program responsible for immunosuppression in tumors. Accordingly, deletion of Irf4 exclusively in Tregs resulted in delayed tumor growth in mice while the abundance of IRF4+ Tregs correlated with poor prognosis in patients with multiple human cancers. Thus, a common mechanism underlies immunosuppression in the tumor microenvironment irrespective of the tumor type.
Keywords: Adaptive immunity; Cancer immunotherapy; Immunology; Oncology; T cells.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- BBS/E/B/000C0409/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/S024468/2/MRC_/Medical Research Council/United Kingdom
- MR/S024468/1/MRC_/Medical Research Council/United Kingdom
- 22597/CRUK_/Cancer Research UK/United Kingdom
- BB/N007794/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
