

Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma
- PMID: 32126144
- PMCID: PMC7065476
- DOI: 10.1182/bloodadvances.2019000779
Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma
Abstract
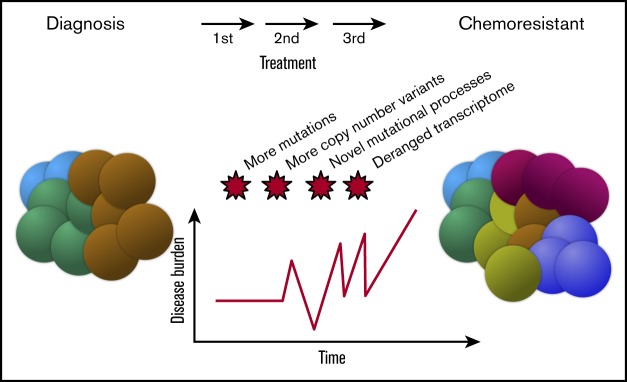
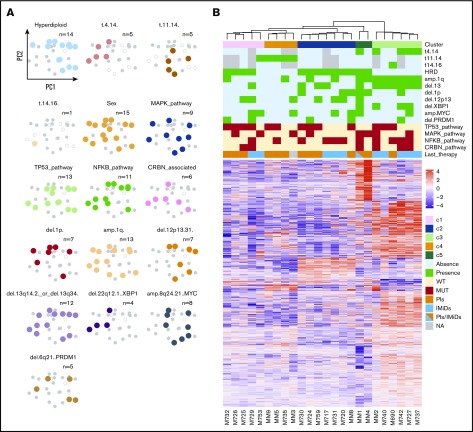
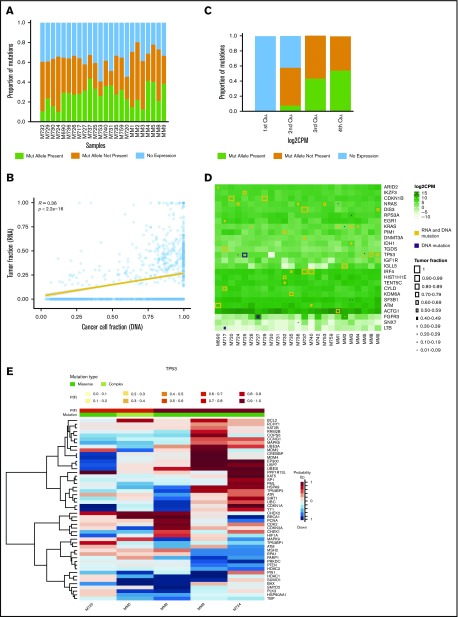
In multiple myeloma, novel treatments with proteasome inhibitors (PIs) and immunomodulatory agents (IMiDs) have prolonged survival but the disease remains incurable. At relapse, next-generation sequencing has shown occasional mutations of drug targets but has failed to identify unifying features that underlie chemotherapy resistance. We studied 42 patients refractory to both PIs and IMiDs. Whole-exome sequencing was performed in 40 patients, and RNA sequencing (RNA-seq) was performed in 27. We found more mutations than were reported at diagnosis and more subclonal mutations, which implies ongoing evolution of the genome of myeloma cells during treatment. The mutational landscape was different from that described in published studies on samples taken at diagnosis. The TP53 pathway was the most frequently inactivated (in 45% of patients). Conversely, point mutations of genes associated with resistance to IMiDs were rare and were always subclonal. Refractory patients were uniquely characterized by having a mutational signature linked to exposure to alkylating agents, whose role in chemotherapy resistance and disease progression remains to be elucidated. RNA-seq analysis showed that treatment or mutations had no influence on clustering, which was instead influenced by karyotypic events. We describe a cluster with both amp(1q) and del(13) characterized by CCND2 upregulation and also overexpression of MCL1, which represents a novel target for experimental treatments. Overall, high-risk features were found in 65% of patients. However, only amp(1q) predicted survival. Gene mutations of IMiD and PI targets are not a preferred mode of drug resistance in myeloma. Chemotherapy resistance of the bulk tumor population is likely attained through differential, yet converging evolution of subclones that are overall variable from patient to patient and within the same patient.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: N.B. received honoraria from Celgene, Amgen, and Janssen. The remaining authors declare no competing financial interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
