

Type II Alexander disease caused by splicing errors and aberrant overexpression of an uncharacterized GFAP isoform
- PMID: 32126152
- PMCID: PMC7491703
- DOI: 10.1002/humu.24008
Type II Alexander disease caused by splicing errors and aberrant overexpression of an uncharacterized GFAP isoform
Erratum in
-
Type II Alexander disease caused by splicing errors and aberrant overexpression of an uncharacterized GFAP isoform.Hum Mutat. 2022 Sep;43(9):1344. doi: 10.1002/humu.24400. Hum Mutat. 2022. PMID: 35920398 Free PMC article. No abstract available.
Abstract
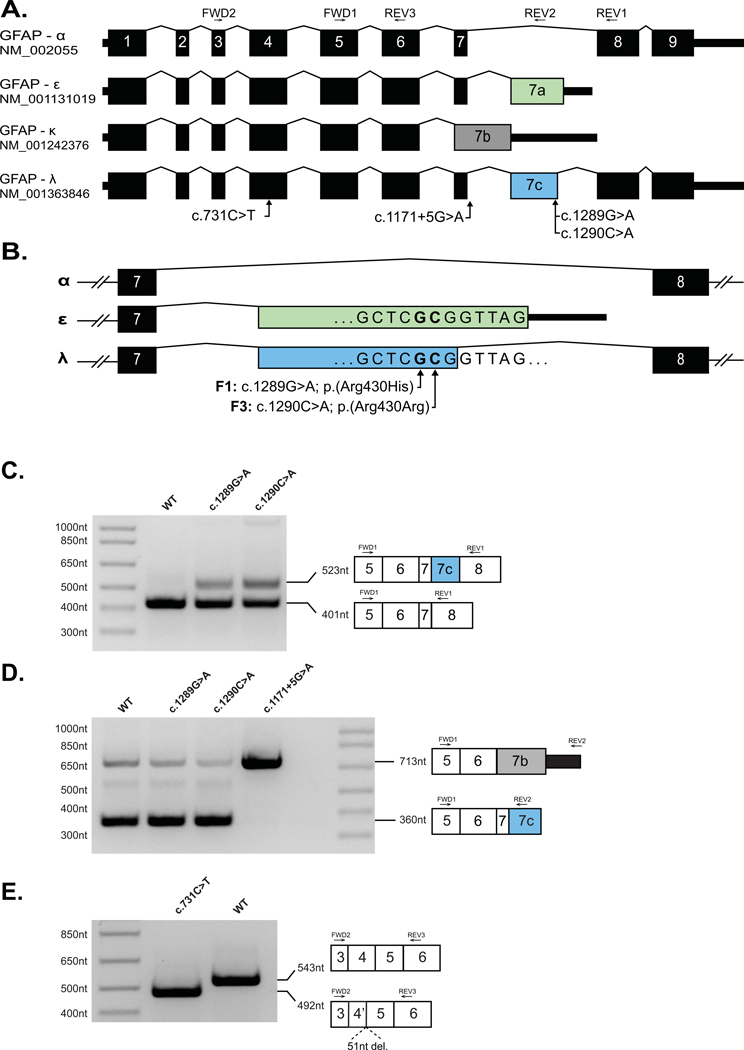
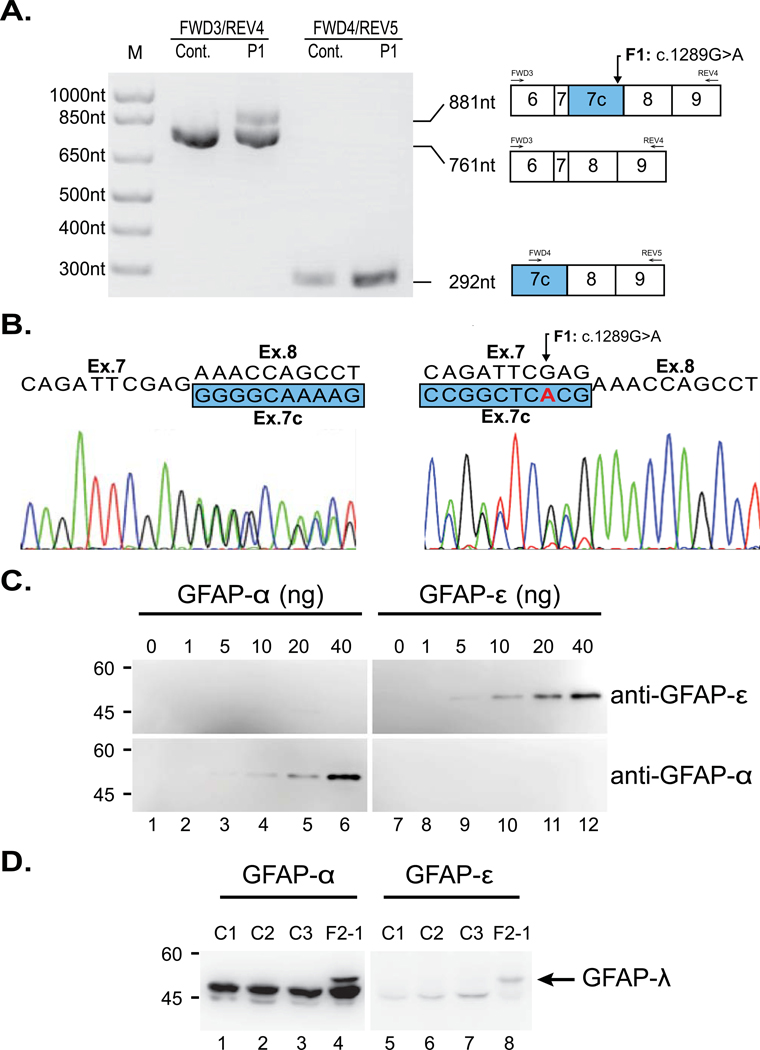
Alexander disease results from gain-of-function mutations in the gene encoding glial fibrillary acidic protein (GFAP). At least eight GFAP isoforms have been described, however, the predominant alpha isoform accounts for ∼90% of GFAP protein. We describe exonic variants identified in three unrelated families with Type II Alexander disease that alter the splicing of GFAP pre-messenger RNA (mRNA) and result in the upregulation of a previously uncharacterized GFAP lambda isoform (NM_001363846.1). Affected members of Family 1 and Family 2 shared the same missense variant, NM_001363846.1:c.1289G>A;p.(Arg430His) while in Family 3 we identified a synonymous variant in the adjacent nucleotide, NM_001363846.1:c.1290C>A;p.(Arg430Arg). Using RNA and protein analysis of brain autopsy samples, and a mini-gene splicing reporter assay, we demonstrate both variants result in the upregulation of the lambda isoform. Our approach demonstrates the importance of characterizing the effect of GFAP variants on mRNA splicing to inform future pathophysiologic and therapeutic study for Alexander disease.
Keywords: Alexander disease; aberrant splicing; leukodystrophy.
© 2020 Wiley Periodicals, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
