

Comprehensive diagnostics of acute myeloid leukemia by whole transcriptome RNA sequencing
- PMID: 32127641
- PMCID: PMC7787979
- DOI: 10.1038/s41375-020-0762-8
Comprehensive diagnostics of acute myeloid leukemia by whole transcriptome RNA sequencing
Abstract
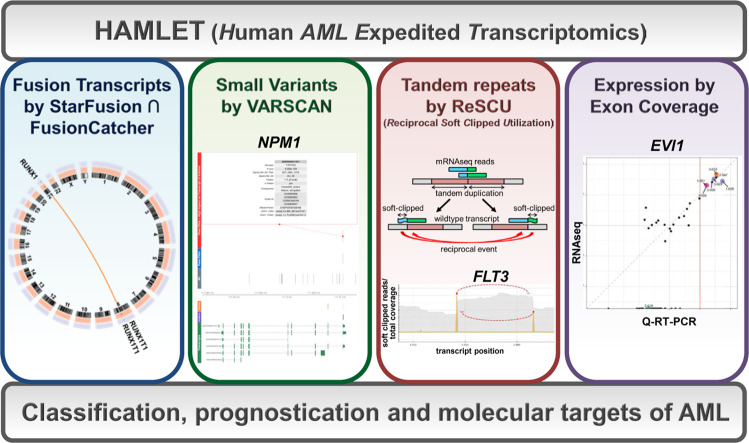
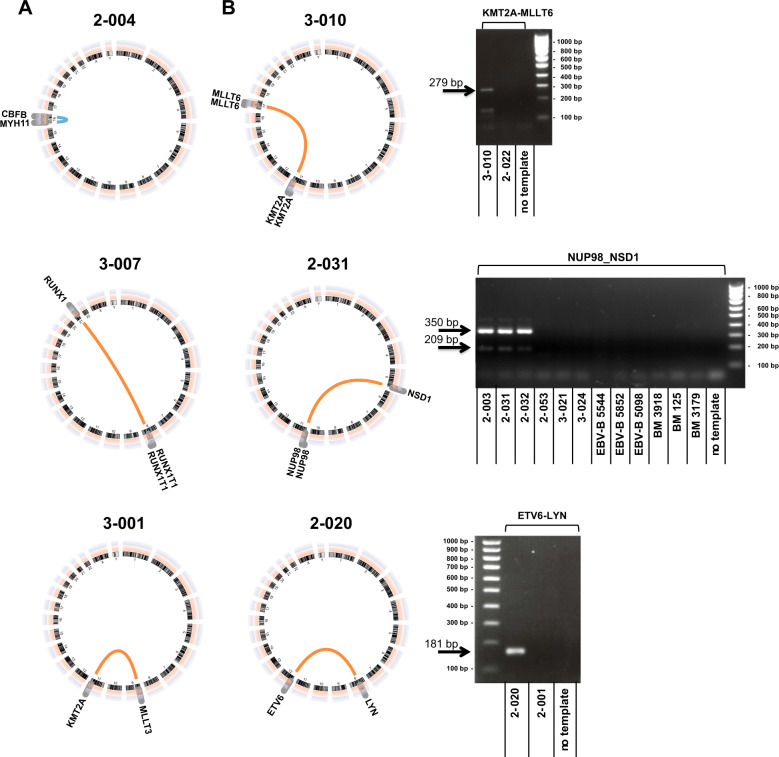
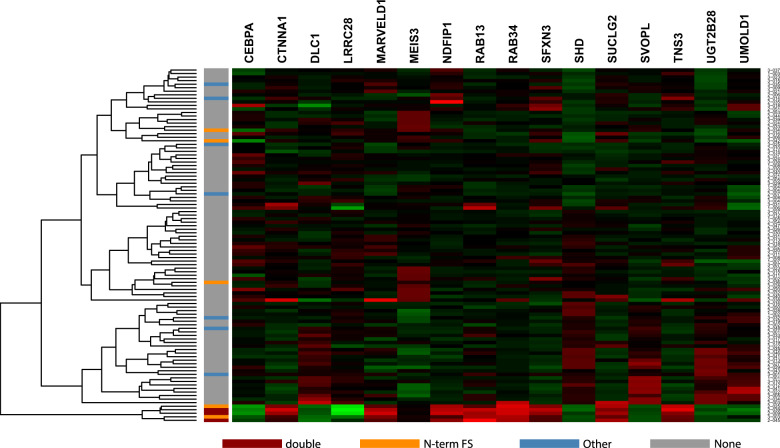
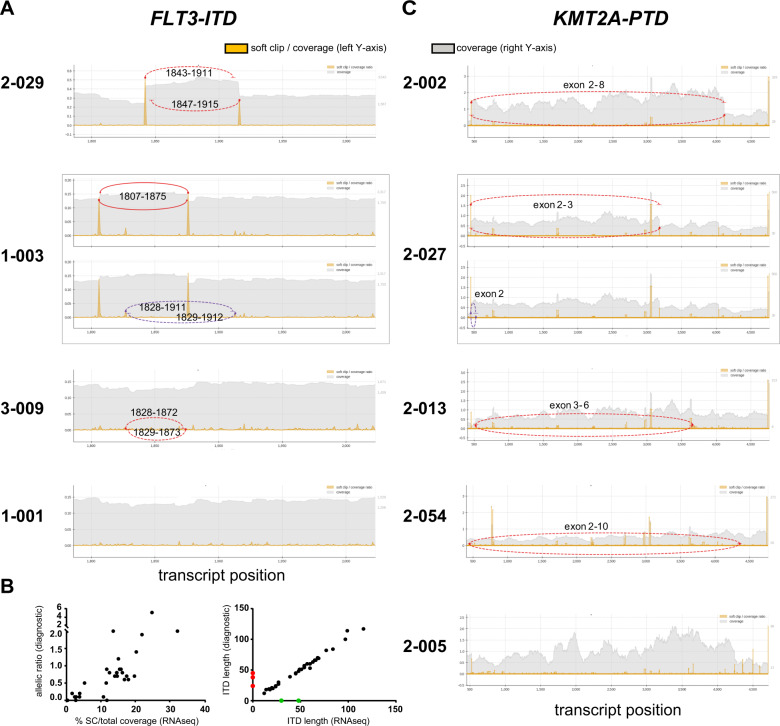
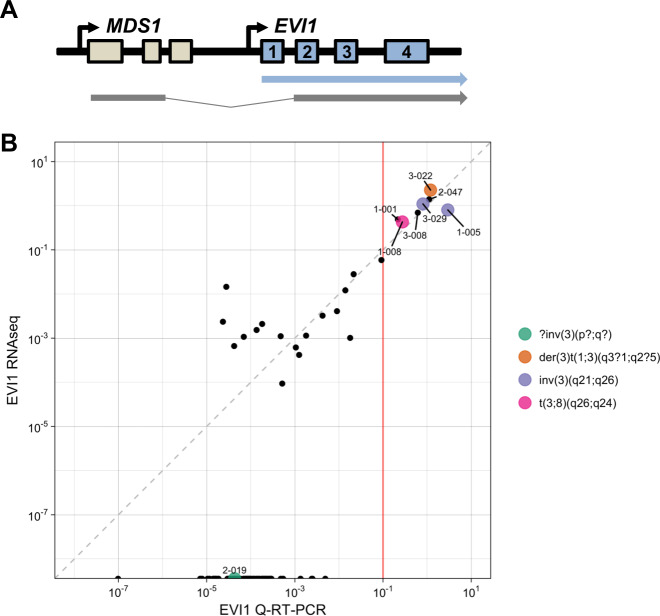
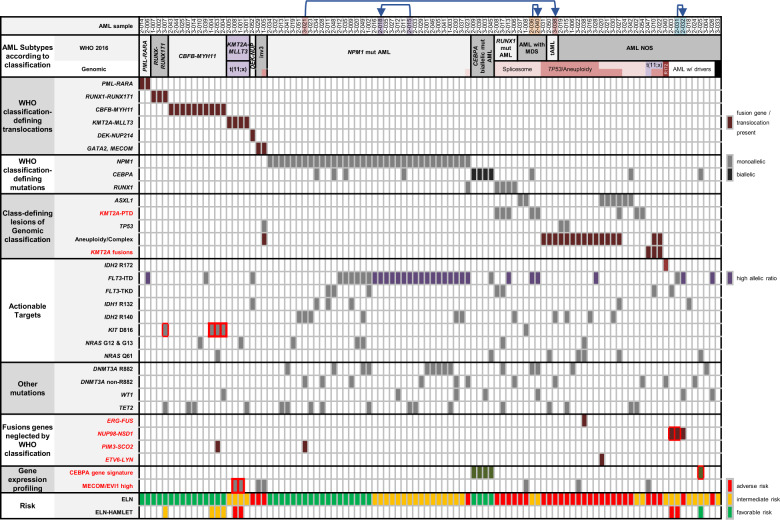
Acute myeloid leukemia (AML) is caused by genetic aberrations that also govern the prognosis of patients and guide risk-adapted and targeted therapy. Genetic aberrations in AML are structurally diverse and currently detected by different diagnostic assays. This study sought to establish whole transcriptome RNA sequencing as single, comprehensive, and flexible platform for AML diagnostics. We developed HAMLET (Human AML Expedited Transcriptomics) as bioinformatics pipeline for simultaneous detection of fusion genes, small variants, tandem duplications, and gene expression with all information assembled in an annotated, user-friendly output file. Whole transcriptome RNA sequencing was performed on 100 AML cases and HAMLET results were validated by reference assays and targeted resequencing. The data showed that HAMLET accurately detected all fusion genes and overexpression of EVI1 irrespective of 3q26 aberrations. In addition, small variants in 13 genes that are often mutated in AML were called with 99.2% sensitivity and 100% specificity, and tandem duplications in FLT3 and KMT2A were detected by a novel algorithm based on soft-clipped reads with 100% sensitivity and 97.1% specificity. In conclusion, HAMLET has the potential to provide accurate comprehensive diagnostic information relevant for AML classification, risk assessment and targeted therapy on a single technology platform.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N. Engl J Med. 2015;373:1136–52. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
