

Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer
- PMID: 32127696
- PMCID: PMC7286536
- DOI: 10.1038/s41574-020-0329-9
Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer
Abstract
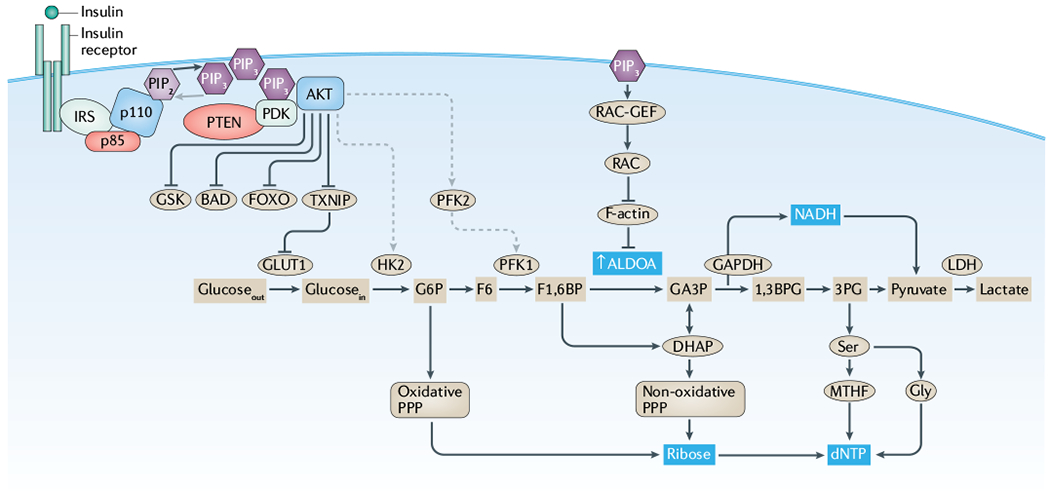
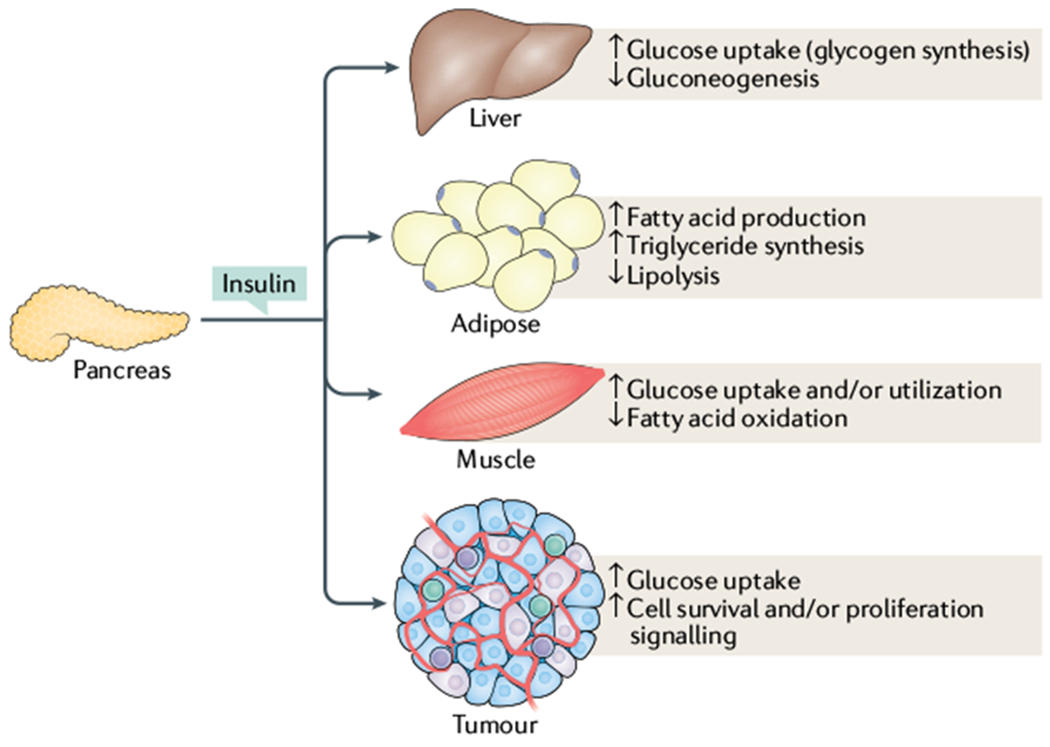
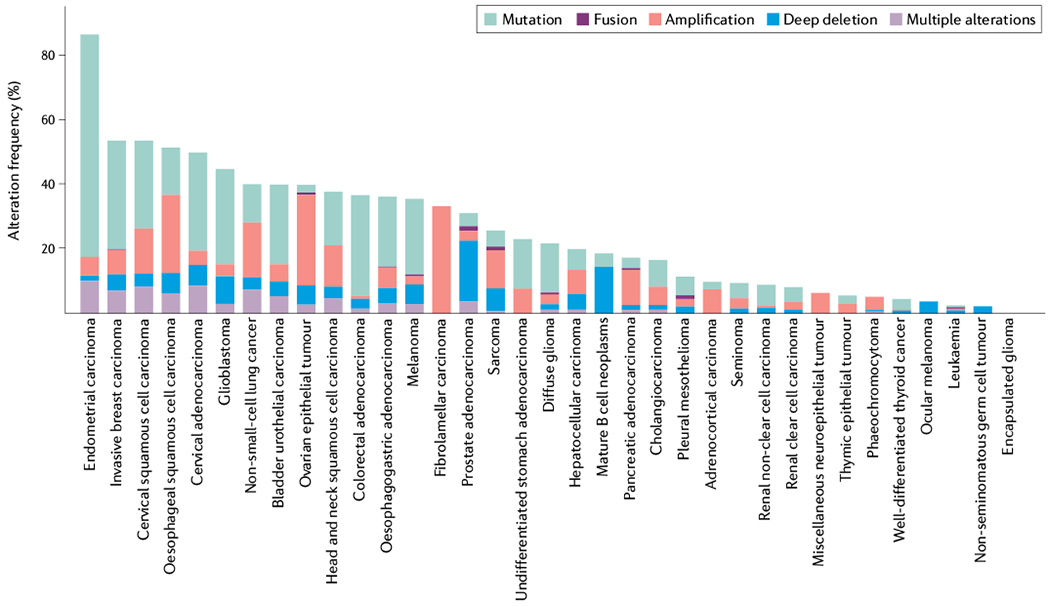
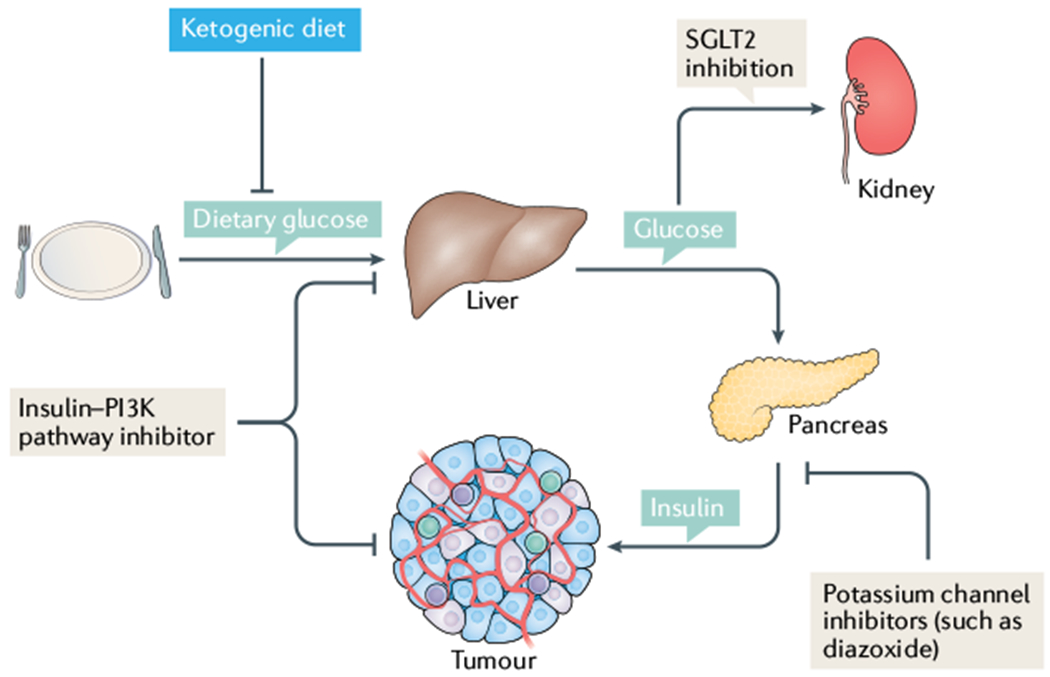
Cancer is driven by incremental changes that accumulate, eventually leading to oncogenic transformation. Although genetic alterations dominate the way cancer biologists think about oncogenesis, growing evidence suggests that systemic factors (for example, insulin, oestrogen and inflammatory cytokines) and their intracellular pathways activate oncogenic signals and contribute to targetable phenotypes. Systemic factors can have a critical role in both tumour initiation and therapeutic responses as increasingly targeted and personalized therapeutic regimens are used to treat patients with cancer. The endocrine system controls cell growth and metabolism by providing extracellular cues that integrate systemic nutrient status with cellular activities such as proliferation and survival via the production of metabolites and hormones such as insulin. When insulin binds to its receptor, it initiates a sequence of phosphorylation events that lead to activation of the catalytic activity of phosphoinositide 3-kinase (PI3K), a lipid kinase that coordinates the intake and utilization of glucose, and mTOR, a kinase downstream of PI3K that stimulates transcription and translation. When chronically activated, the PI3K pathway can drive malignant transformation. Here, we discuss the insulin-PI3K signalling cascade and emphasize its roles in normal cells (including coordinating cell metabolism and growth), highlighting the features of this network that make it ideal for co-option by cancer cells. Furthermore, we discuss how this signalling network can affect therapeutic responses and how novel metabolic-based strategies might enhance treatment efficacy for cancer.
Conflict of interest statement
Competing interests
B.D.H., M.D.G. and L.C.C. are all founders of and consultants for Faeth, a company developing nutrition for cancer care. L.C.C. is a founder and member of the scientific advisory board and board of directors of Agios and Petra Pharma, which are companies developing drugs to target metabolism.
Figures
References
-
- Karamitsos DT The story of insulin discovery. Diabetes Res. Clin. Pract 93, S2–S8 (2011). - PubMed
-
- Banting FG & Best CH Pancreatic Extracts (Toronto Univ. Library, 1922).
-
- Pollak M Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 (2008). - PubMed
-
- Dong MQ et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 317, 660–663 (2007). - PubMed
-
- Engelman JA, Luo J & Cantley LC The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet 7, 606–619 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
