

Influence of EGFR-activating mutations on sensitivity to tyrosine kinase inhibitors in a KRAS mutant non-small cell lung cancer cell line
- PMID: 32130260
- PMCID: PMC7055889
- DOI: 10.1371/journal.pone.0229712
Influence of EGFR-activating mutations on sensitivity to tyrosine kinase inhibitors in a KRAS mutant non-small cell lung cancer cell line
Abstract
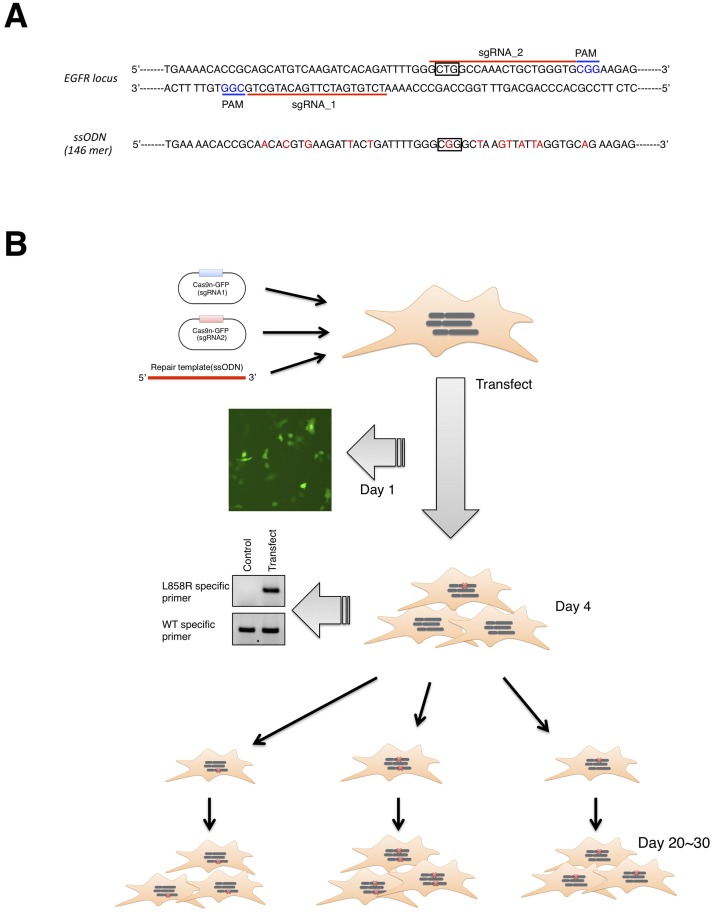
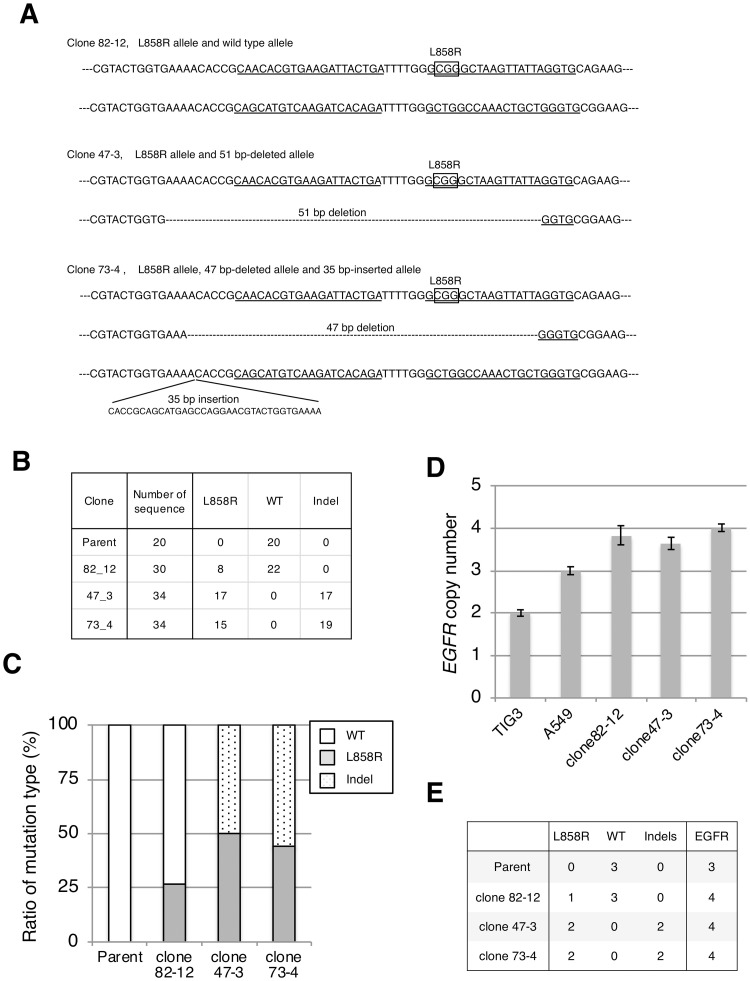
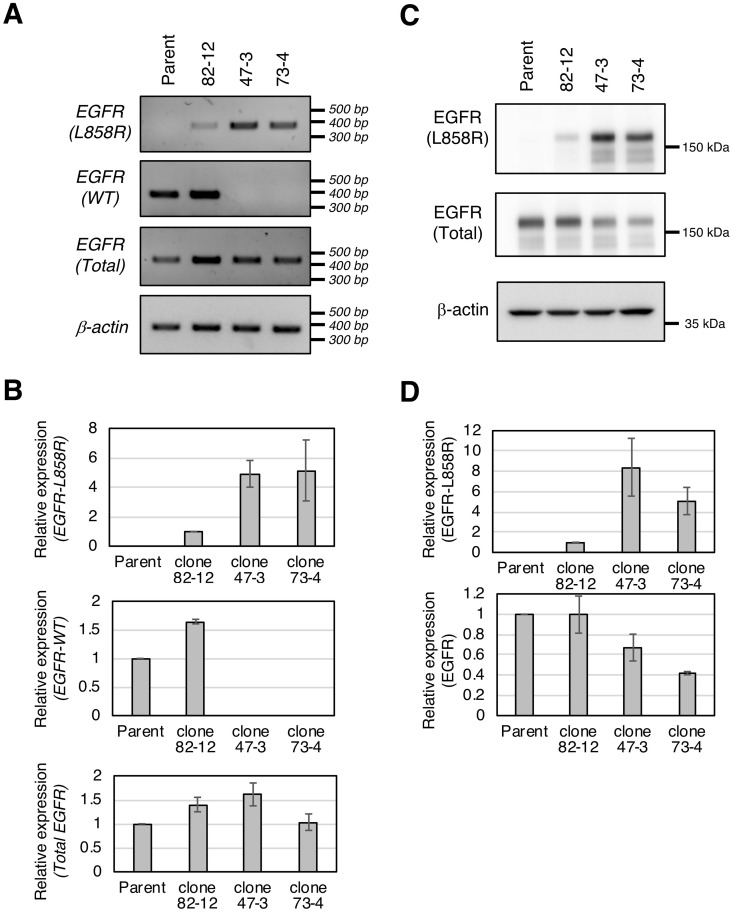
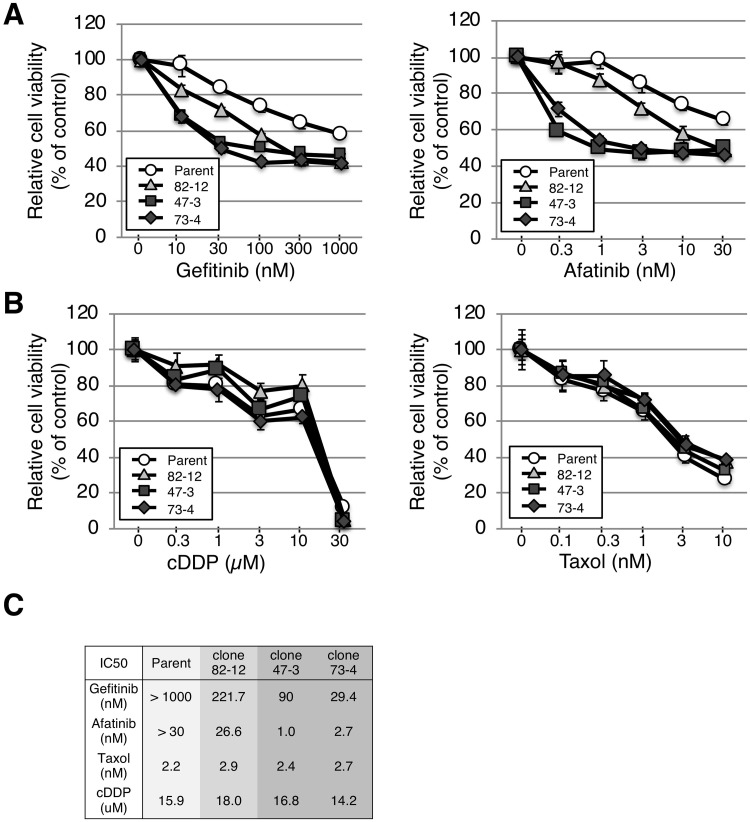
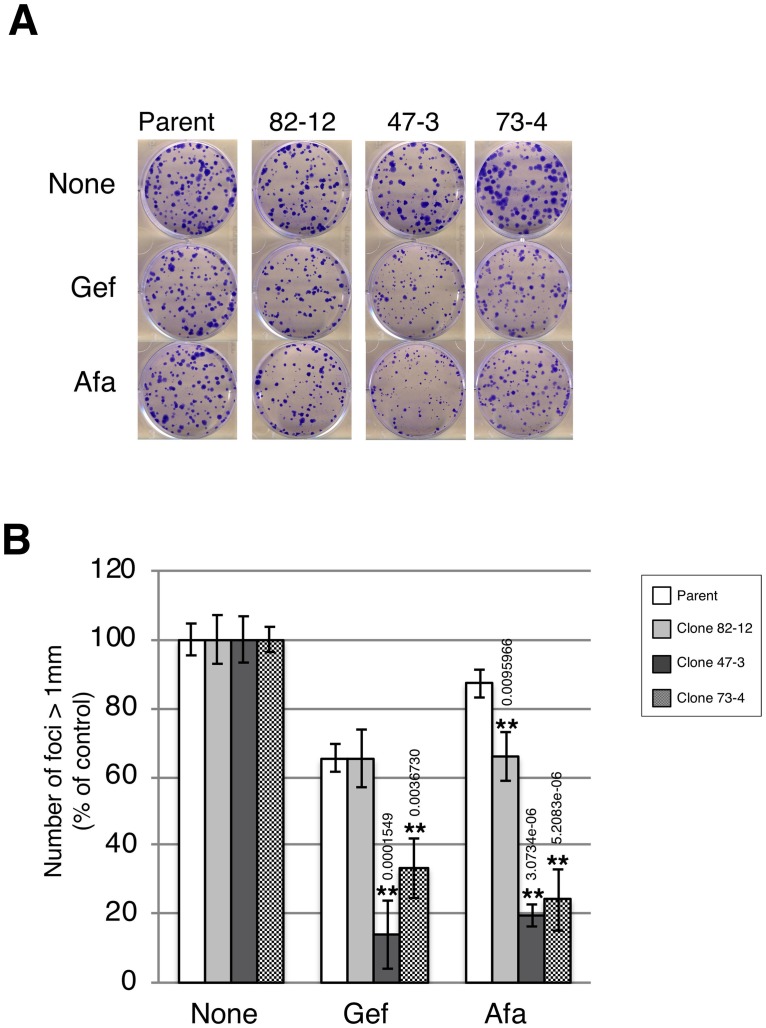
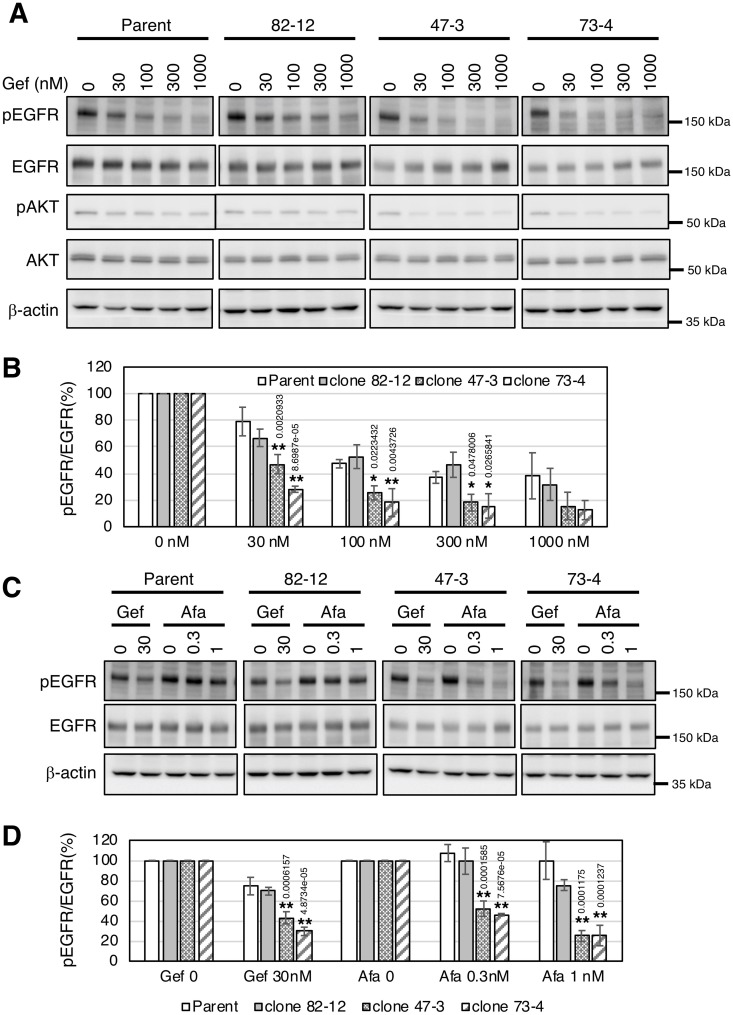
In non-small cell lung cancer (NSCLC), oncogenic driver mutations including those in KRAS and EGFR are typically mutually exclusive. However, recent reports indicate that multiple driver mutations are found in a certain percentage of cancers, and that the therapeutic responses of such cases with co-mutations of driver genes are largely unclear. Here, using CRISPR-Cas9-mediated genome editing, we generated isogenic cell lines harboring one or two copies of an EGFR-activating mutation from the human NSCLC cell line A549, which is known to harbor a homozygous KRAS gene mutation. In comparison with parent cells with KRAS mutation alone, cells with concomitant EGFR mutation exhibited higher sensitivity to EGFR-tyrosine kinase inhibitors (TKIs) but not to conventional anti-cancer drugs. In particular, cells with two copies of EGFR mutation were markedly more sensitive to EGFR-TKIs compared with parent cells. Thus, the presence of concomitant EGFR mutation can affect the TKI response of KRAS-mutated cells, implying that EGFR-TKI may represent an effective treatment option against NSCLC with EGFR/KRAS co-mutation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of oncology: official journal of the European Society for Medical Oncology. 2013;24(9):2371–6. Epub 2013/06/01. 10.1093/annonc/mdt205 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
