

Dynamics of Co-translational Membrane Protein Integration and Translocation via the Sec Translocon
- PMID: 32130863
- PMCID: PMC7338273
- DOI: 10.1021/jacs.9b07820
Dynamics of Co-translational Membrane Protein Integration and Translocation via the Sec Translocon
Abstract
An important aspect of cellular function is the correct targeting and delivery of newly synthesized proteins. Central to this task is the machinery of the Sec translocon, a transmembrane channel that is involved in both the translocation of nascent proteins across cell membranes and the integration of proteins into the membrane. Considerable experimental and computational effort has focused on the Sec translocon and its role in nascent protein biosynthesis, including the correct folding and expression of integral membrane proteins. However, the use of molecular simulation methods to explore Sec-facilitated protein biosynthesis is hindered by the large system sizes and long (i.e., minute) time scales involved. In this work, we describe the development and application of a coarse-grained simulation approach that addresses these challenges and allows for direct comparison with both in vivo and in vitro experiments. The method reproduces a wide range of experimental observations, providing new insights into the underlying molecular mechanisms, predictions for new experiments, and a strategy for the rational enhancement of membrane protein expression levels.
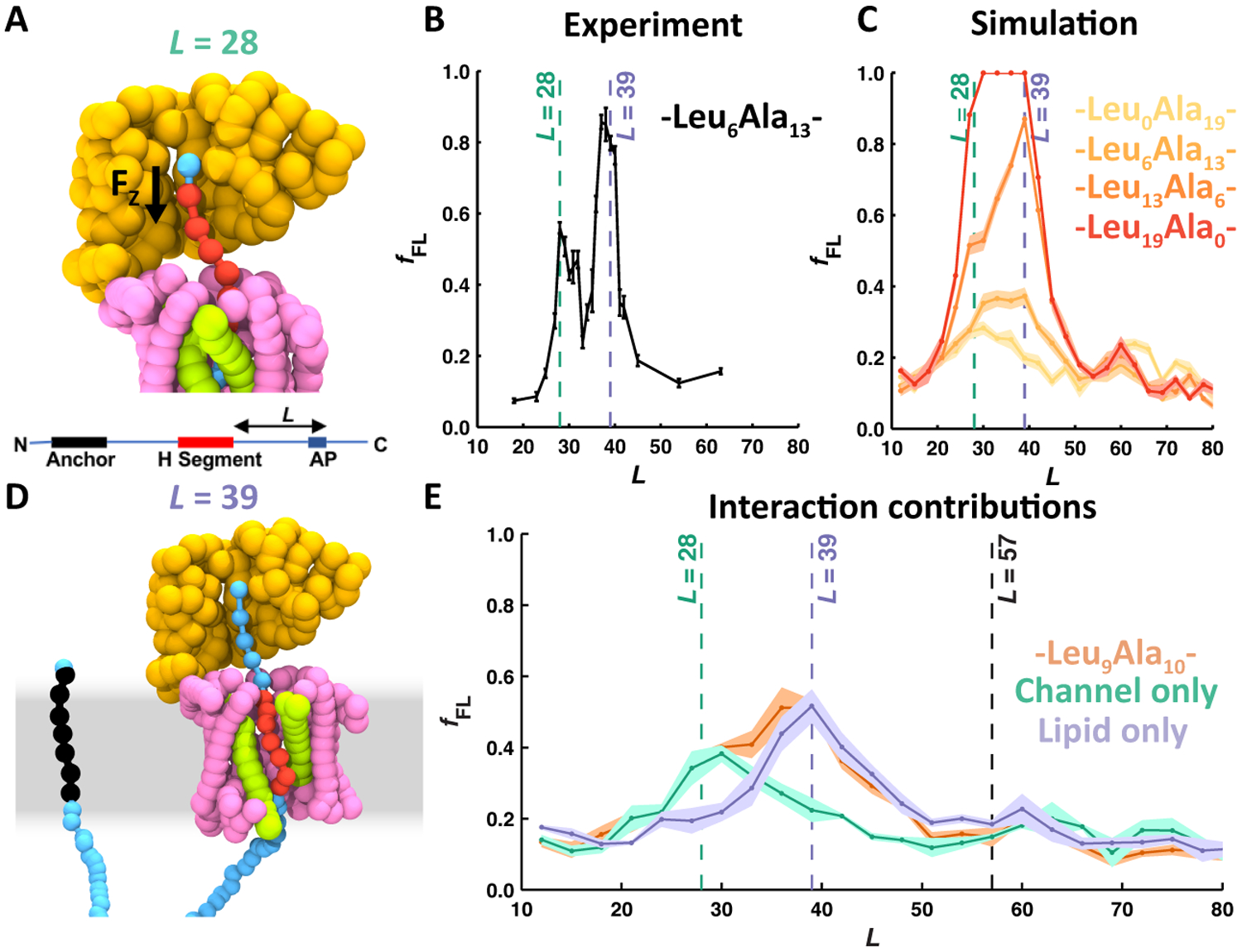
Figures






Similar articles
-
Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes.Int J Mol Sci. 2021 Oct 2;22(19):10705. doi: 10.3390/ijms221910705. Int J Mol Sci. 2021. PMID: 34639046 Free PMC article. Review.
-
Structurally detailed coarse-grained model for Sec-facilitated co-translational protein translocation and membrane integration.PLoS Comput Biol. 2017 Mar 22;13(3):e1005427. doi: 10.1371/journal.pcbi.1005427. eCollection 2017 Mar. PLoS Comput Biol. 2017. PMID: 28328943 Free PMC article.
-
Forces on Nascent Polypeptides during Membrane Insertion and Translocation via the Sec Translocon.Biophys J. 2018 Nov 20;115(10):1885-1894. doi: 10.1016/j.bpj.2018.10.002. Epub 2018 Oct 10. Biophys J. 2018. PMID: 30366631 Free PMC article.
-
Structure and Dynamics of the Central Lipid Pool and Proteins of the Bacterial Holo-Translocon.Biophys J. 2019 May 21;116(10):1931-1940. doi: 10.1016/j.bpj.2019.04.002. Epub 2019 Apr 9. Biophys J. 2019. PMID: 31053257 Free PMC article.
-
Structural Basis of the Sec Translocon and YidC Revealed Through X-ray Crystallography.Protein J. 2019 Jun;38(3):249-261. doi: 10.1007/s10930-019-09830-x. Protein J. 2019. PMID: 30972527 Review.
Cited by
-
Facade-Based Bicelles as a New Tool for Production of Active Membrane Proteins in a Cell-Free System.Int J Mol Sci. 2023 Oct 3;24(19):14864. doi: 10.3390/ijms241914864. Int J Mol Sci. 2023. PMID: 37834312 Free PMC article.
-
Guardians of the Cell: State-of-the-Art of Membrane Proteins from a Computational Point-of-View.Methods Mol Biol. 2021;2315:3-28. doi: 10.1007/978-1-0716-1468-6_1. Methods Mol Biol. 2021. PMID: 34302667
-
Folding and Insertion of Transmembrane Helices at the ER.Int J Mol Sci. 2021 Nov 26;22(23):12778. doi: 10.3390/ijms222312778. Int J Mol Sci. 2021. PMID: 34884581 Free PMC article. Review.
-
Coordination of -1 programmed ribosomal frameshifting by transcript and nascent chain features revealed by deep mutational scanning.Nucleic Acids Res. 2021 Dec 16;49(22):12943-12954. doi: 10.1093/nar/gkab1172. Nucleic Acids Res. 2021. PMID: 34871407 Free PMC article.
-
Molecular Modeling of Signal Peptide Recognition by Eukaryotic Sec Complexes.Int J Mol Sci. 2021 Oct 2;22(19):10705. doi: 10.3390/ijms221910705. Int J Mol Sci. 2021. PMID: 34639046 Free PMC article. Review.
References
-
- Rapoport TA Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. - PubMed
-
- White SH; von Heijne G How translocons select transmembrane helices. Annu. Rev. Biophys 2008, 37, 23–42. - PubMed
-
- von Heijne G Membrane-protein topology. Nat. Rev. Mol. Cell Biol 2006, 7, 909–918. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
