

Review
doi: 10.2215/CJN.09480819.
Epub 2020 Mar 4.
The Changing Landscape of Fabry Disease
Affiliations
- PMID: 32132142
- PMCID: PMC7133143
- DOI: 10.2215/CJN.09480819
Item in Clipboard
Review
The Changing Landscape of Fabry Disease
Clin J Am Soc Nephrol.
.
No abstract available
Keywords: Fabry disease; Fabry’s disease; adult; agalsidase α; agalsidase β; alphagalactosidase; angiokeratoma; cardiac myocytes; cardiovascular disease; cause of death; enzyme replacement therapy; female; follow-up studies; genetic renal disease; humans; kidney biopsy; kidney transplantation; longevity; male; podocyte; progression of renal failure; renal dialysis; renal function decline; vascular disease; vascular endothelium.
Figures
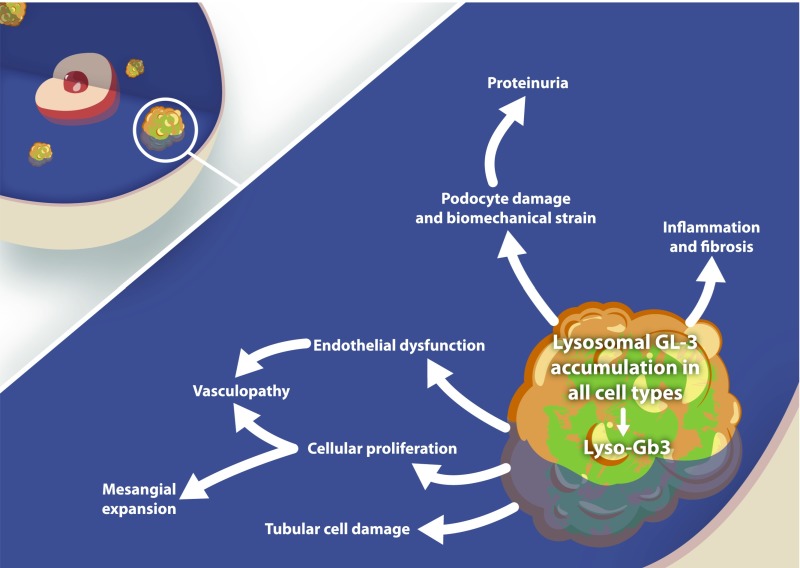
Pathomechanisms of Fabry nephropathy [Eikrem et al. (81)]. GL3, globotriaocylceramide; Lyso-Gb3, globotriaosylsphingosine.
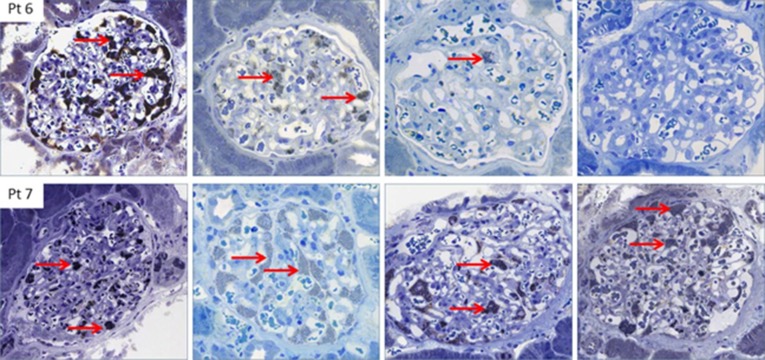
Histology of Fabry nephropathy. Dose-dependent clearance of podocyte GL3 (red arrows) in two brothers with classic Fabry disease, aged 13 and 15 years at initiation of enzyme replacement therapy. Biopsies are baseline (upper and lower left column, respectively), and after 3, 7, and 13 years of agalsidase therapy. After 6 years of enzyme replacement therapy, the younger brother (upper row) was switched to agalsidase β 1.0 mg/kg every other week. After the switch, the podocytes were virtually completely cleared of GL3, whereas his older brother (lower row), who received a lower cumulative agalsidase dose, continued to have a full podocyte score after a total of 13 years [Skrunes et al. (41)]. Pt, patient.
References
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L: Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med 276: 1163–1167, 1967 - PubMed
-
- Anderson W: A case of “angeiokeratoma”. Br J Dermatol 10: 113–117, 1898
-
- Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR: Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 138: 338–346, 2003 - PubMed
-
- Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin Iii HA, Kopp JB: Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81: 122–138, 2002 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
