

The Transcriptional Cofactor VGLL1 Drives Transcription of Human Papillomavirus Early Genes via TEAD1
- PMID: 32132238
- PMCID: PMC7199408
- DOI: 10.1128/JVI.01945-19
The Transcriptional Cofactor VGLL1 Drives Transcription of Human Papillomavirus Early Genes via TEAD1
Abstract
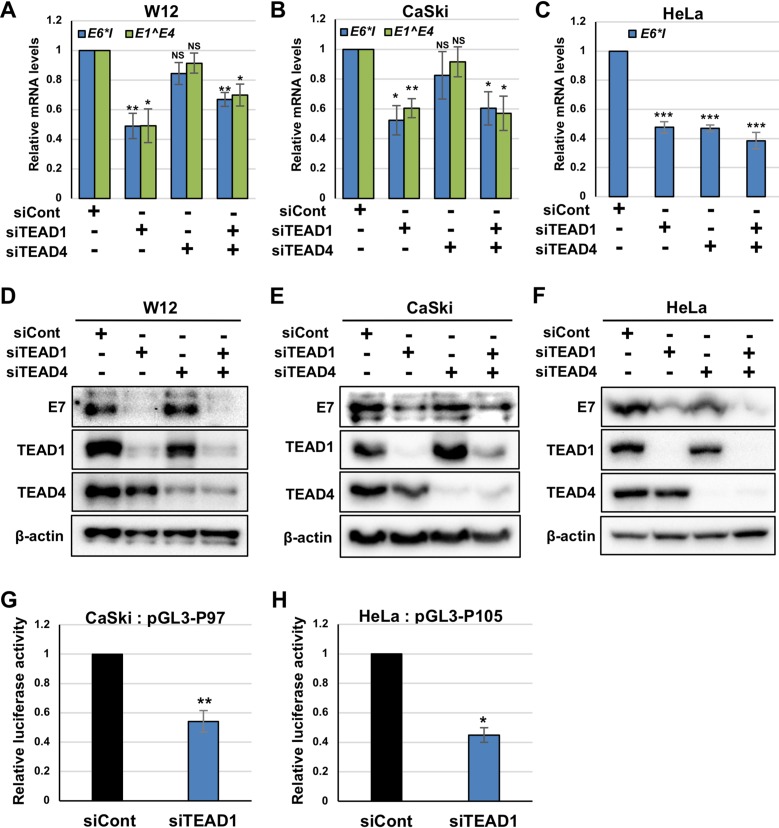
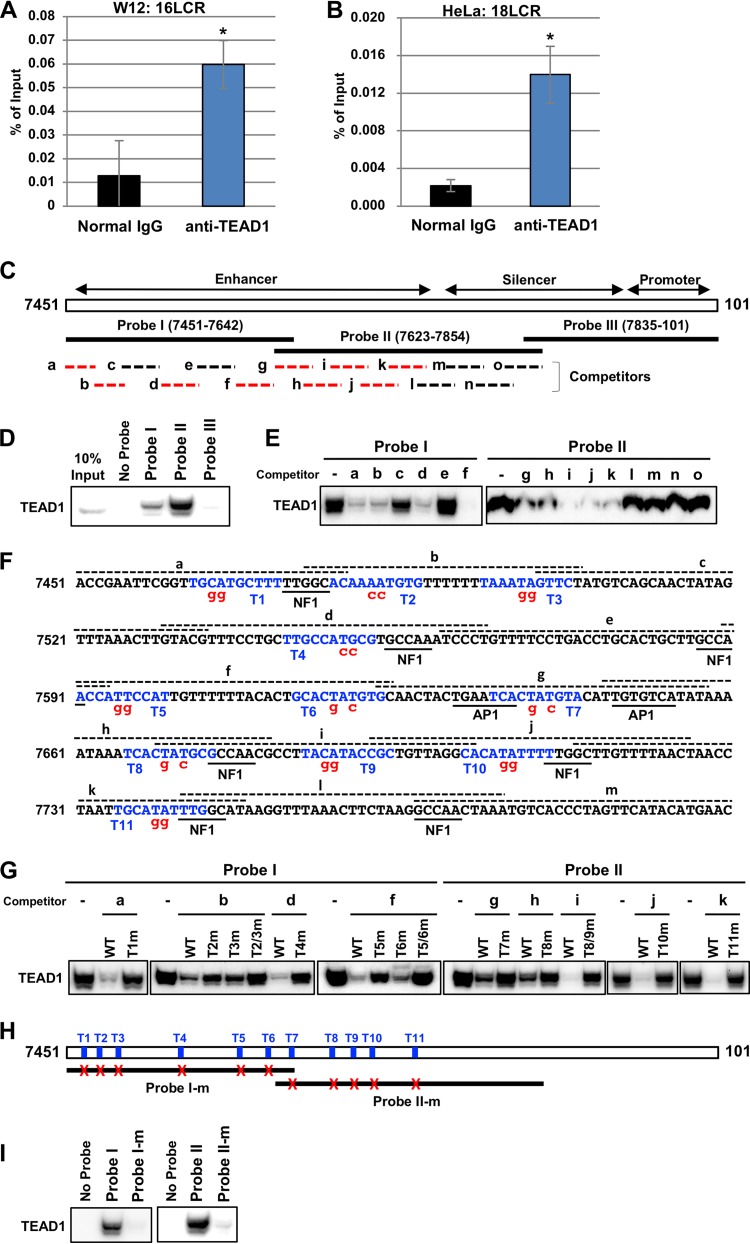
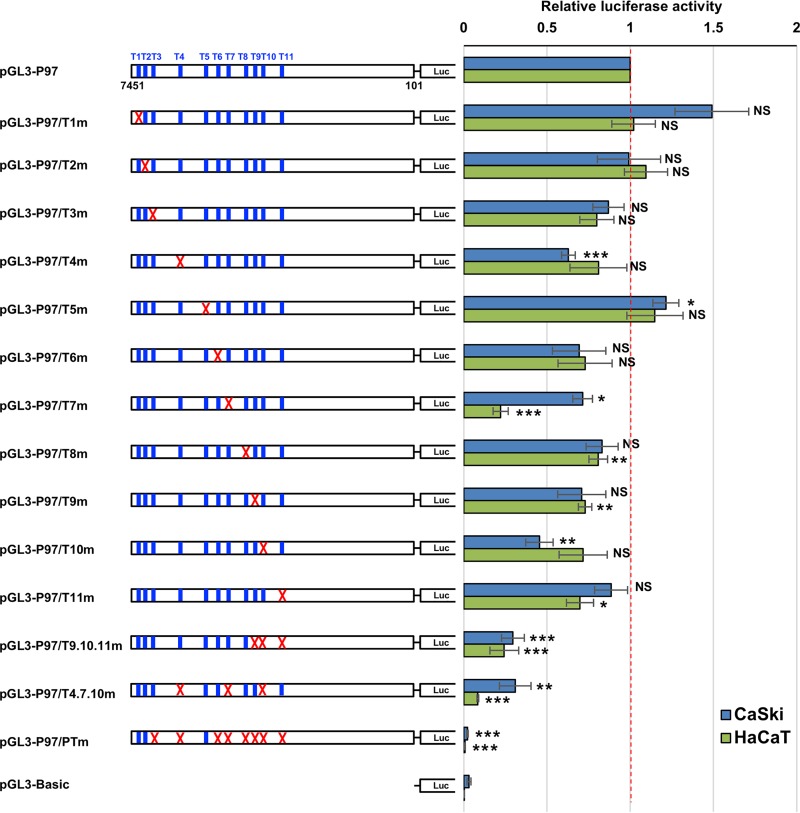
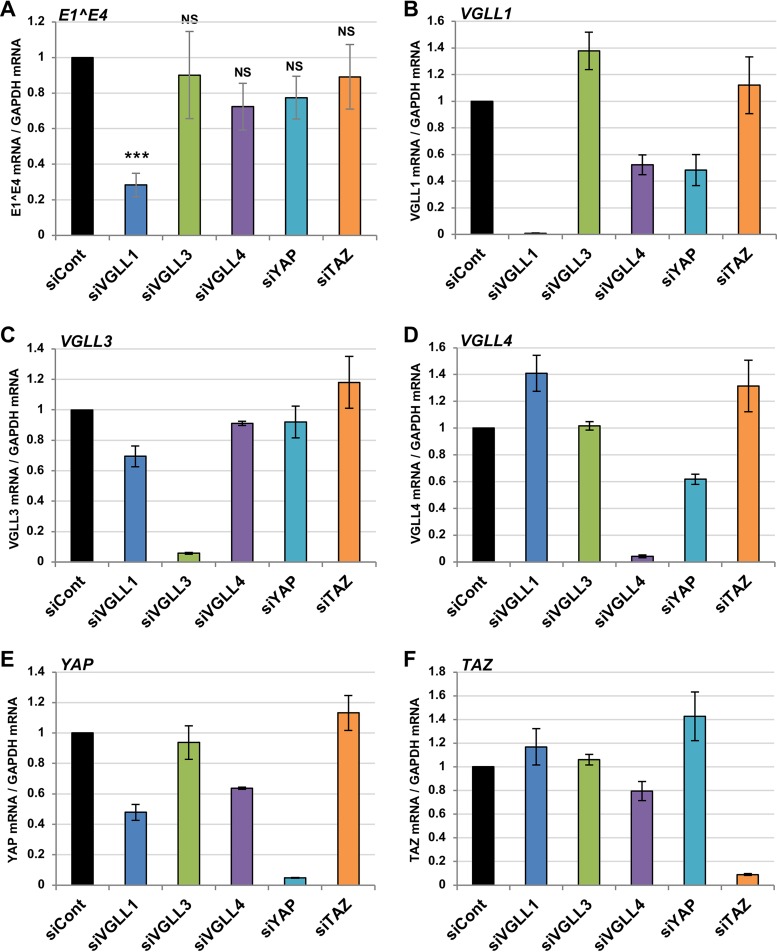
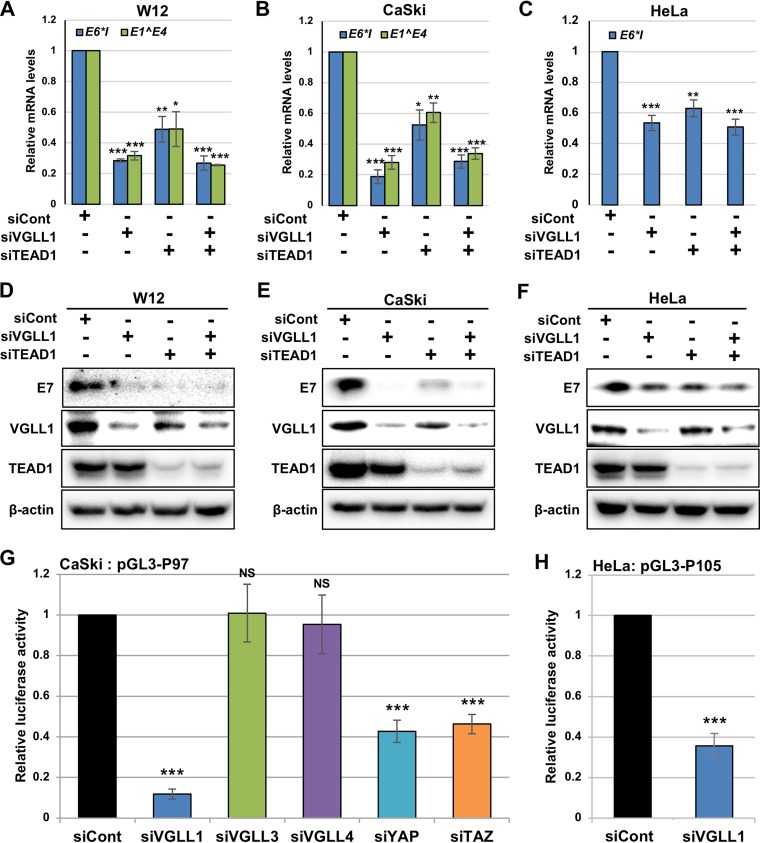
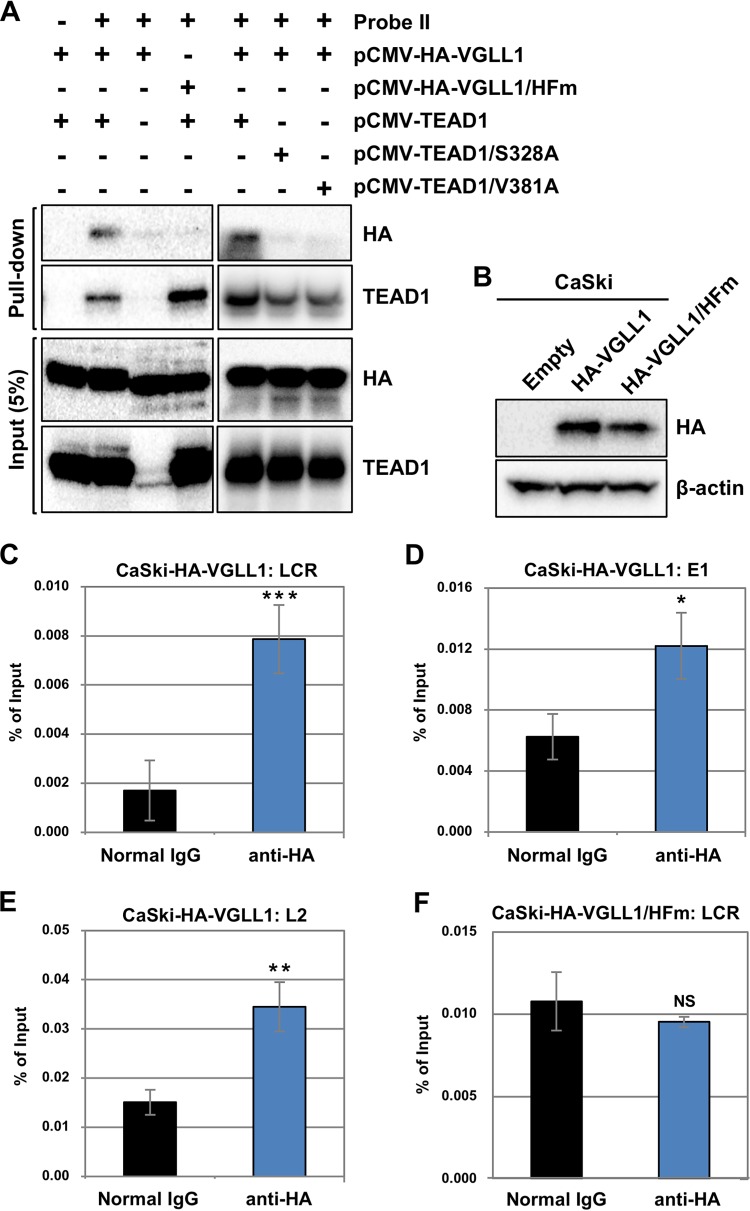
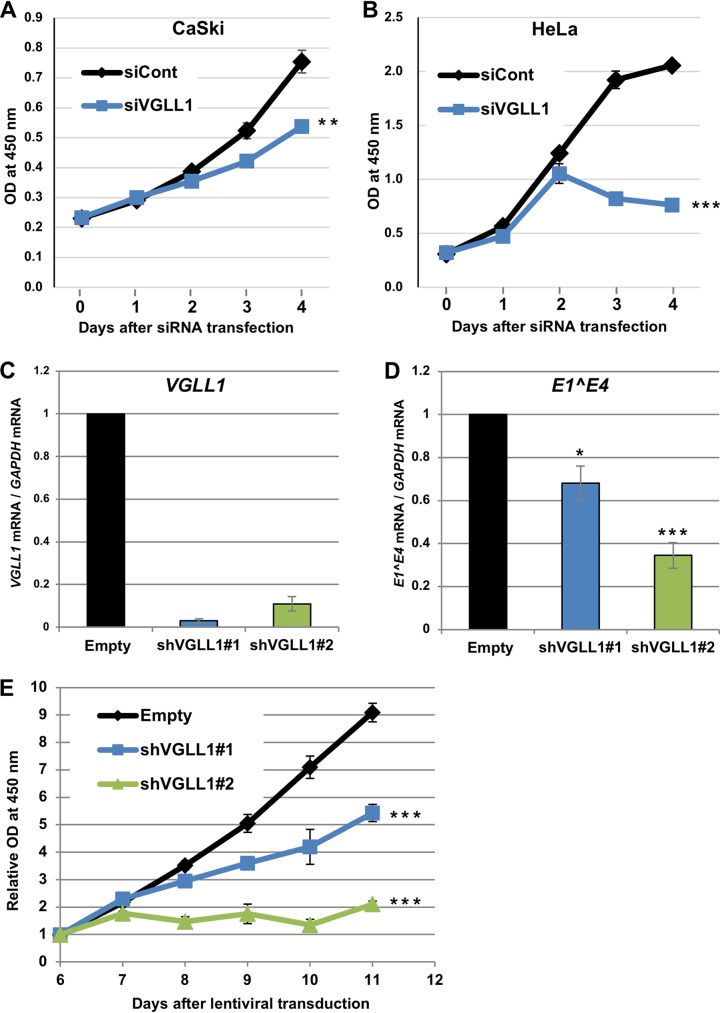
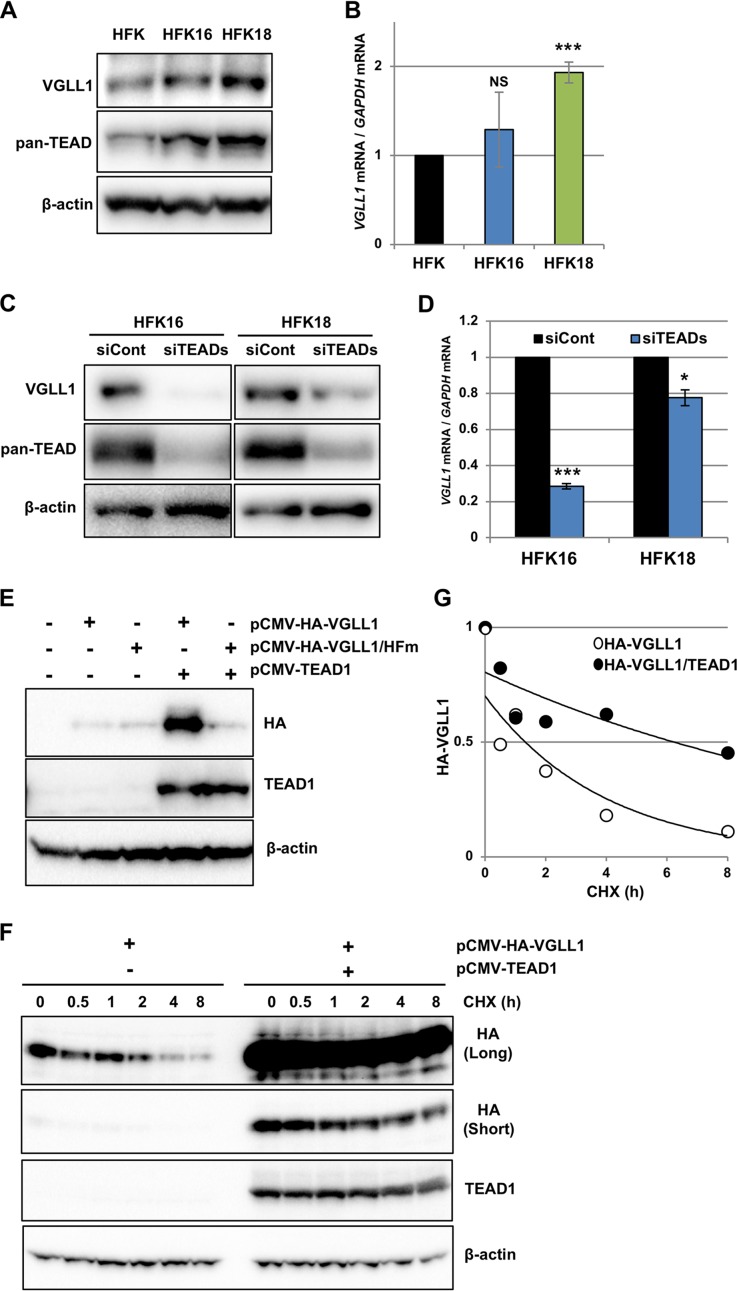
The TEAD family of transcription factors requires associating cofactors to induce gene expression. TEAD1 is known to activate the early promoter of human papillomavirus (HPV), but the precise mechanisms of TEAD1-mediated transactivation of the HPV promoter, including its relevant cofactors, remain unexplored. Here, we reveal that VGLL1, a TEAD-interacting cofactor, contributes to HPV early gene expression. Knockdown of VGLL1 and/or TEAD1 led to a decrease in viral early gene expression in human cervical keratinocytes and cervical cancer cell lines. We identified 11 TEAD1 target sites in the HPV16 long control region (LCR) by in vitro DNA pulldown assays; 8 of these sites contributed to the transcriptional activation of the early promoter in luciferase reporter assays. VGLL1 bound to the HPV16 LCR via its interaction with TEAD1 both in vitro and in vivo Furthermore, introducing HPV16 and HPV18 whole genomes into primary human keratinocytes led to increased levels of VGLL1, due in part to the upregulation of TEADs. These results suggest that multiple VGLL1/TEAD1 complexes are recruited to the LCR to support the efficient transcription of HPV early genes.IMPORTANCE Although a number of transcription factors have been reported to be involved in HPV gene expression, little is known about the cofactors that support HPV transcription. In this study, we demonstrate that the transcriptional cofactor VGLL1 plays a prominent role in HPV early gene expression, dependent on its association with the transcription factor TEAD1. Whereas TEAD1 is ubiquitously expressed in a variety of tissues, VGLL1 displays tissue-specific expression and is implicated in the development and differentiation of epithelial lineage tissues, where HPV gene expression occurs. Our results suggest that VGLL1 may contribute to the epithelial specificity of HPV gene expression, providing new insights into the mechanisms that regulate HPV infection. Further, VGLL1 is also critical for the growth of cervical cancer cells and may represent a novel therapeutic target for HPV-associated cancers.
Keywords: HPV; gene expression; transcription cofactor; transcription factor.
Copyright © 2020 American Society for Microbiology.
Figures
Similar articles
-
Nuclear proinflammatory cytokine S100A9 enhances expression of human papillomavirus oncogenes via transcription factor TEAD1.J Virol. 2023 Aug 31;97(8):e0081523. doi: 10.1128/jvi.00815-23. Epub 2023 Aug 14. J Virol. 2023. PMID: 37578237 Free PMC article.
-
Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor.J Virol. 2017 Feb 28;91(6):e02413-16. doi: 10.1128/JVI.02413-16. Print 2017 Mar 15. J Virol. 2017. PMID: 28077648 Free PMC article.
-
[Possible role of transcription factor AP1 in the tissue-specific regulation of human papillomavirus].Rev Invest Clin. 2002 May-Jun;54(3):231-42. Rev Invest Clin. 2002. PMID: 12183893 Review. Spanish.
-
Transcriptional activation of the human papillomavirus type 5 and 16 long control region in cells from cutaneous and mucosal origin.Virol J. 2007 Mar 12;4:27. doi: 10.1186/1743-422X-4-27. Virol J. 2007. PMID: 17352804 Free PMC article.
-
Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma.Virology. 2009 Feb 20;384(2):375-9. doi: 10.1016/j.virol.2008.11.014. Epub 2008 Dec 7. Virology. 2009. PMID: 19064276 Review.
Cited by
-
Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter.Cancers (Basel). 2021 Sep 15;13(18):4613. doi: 10.3390/cancers13184613. Cancers (Basel). 2021. PMID: 34572841 Free PMC article.
-
YAP/TAZ-TEAD activity promotes the malignant transformation of cervical intraepithelial neoplasia through enhancing the characteristics and Warburg effect of cancer stem cells.Apoptosis. 2024 Aug;29(7-8):1198-1210. doi: 10.1007/s10495-023-01935-0. Epub 2024 Mar 29. Apoptosis. 2024. PMID: 38553612 Free PMC article.
-
Therapeutic vaccine-mediated Gag-specific CD8+ T-cell induction under anti-retroviral therapy augments anti-virus efficacy of CD8+ cells in simian immunodeficiency virus-infected macaques.Sci Rep. 2020 Jul 9;10(1):11394. doi: 10.1038/s41598-020-68267-w. Sci Rep. 2020. PMID: 32647227 Free PMC article.
-
Multiple Roles of Vestigial-Like Family Members in Tumor Development.Front Oncol. 2020 Jul 24;10:1266. doi: 10.3389/fonc.2020.01266. eCollection 2020. Front Oncol. 2020. PMID: 32793503 Free PMC article. Review.
-
Nuclear proinflammatory cytokine S100A9 enhances expression of human papillomavirus oncogenes via transcription factor TEAD1.J Virol. 2023 Aug 31;97(8):e0081523. doi: 10.1128/jvi.00815-23. Epub 2023 Aug 14. J Virol. 2023. PMID: 37578237 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
