

Differential DNA methylation of vocal and facial anatomy genes in modern humans
- PMID: 32132541
- PMCID: PMC7055320
- DOI: 10.1038/s41467-020-15020-6
Differential DNA methylation of vocal and facial anatomy genes in modern humans
Abstract
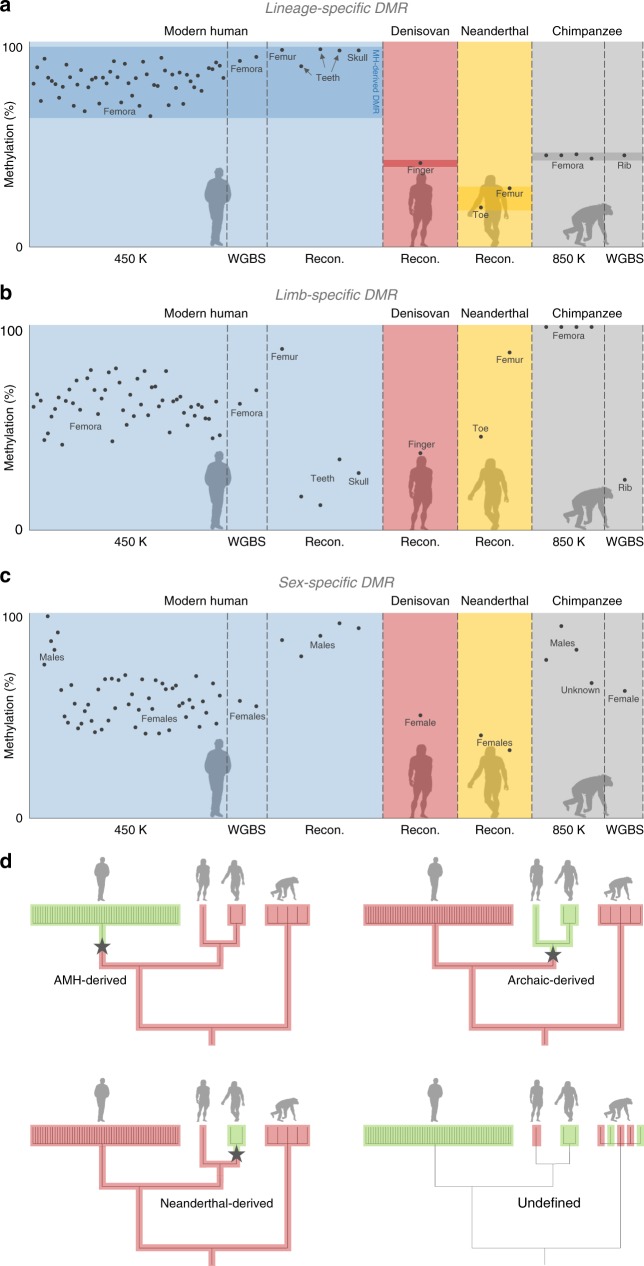
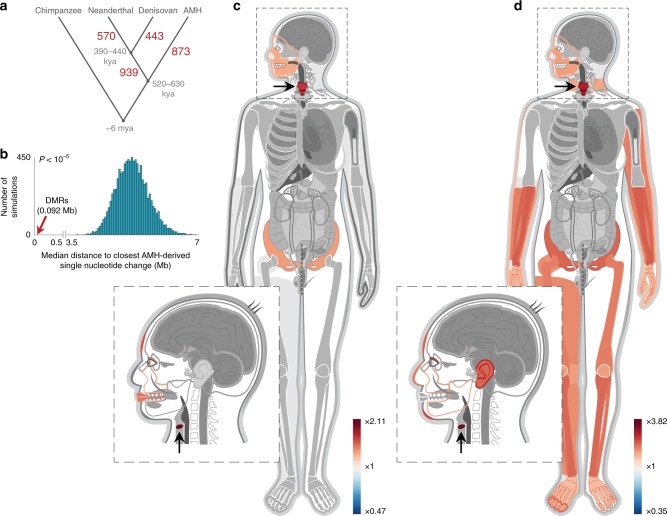
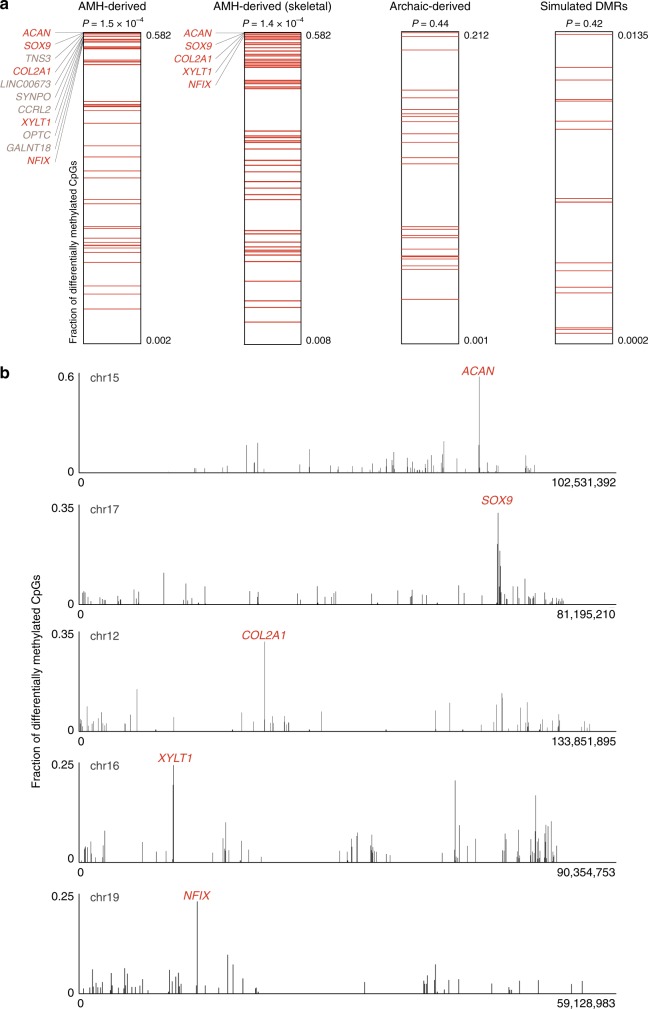
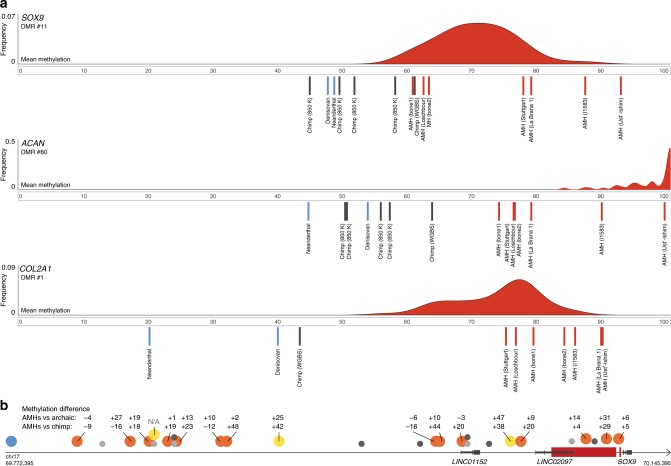
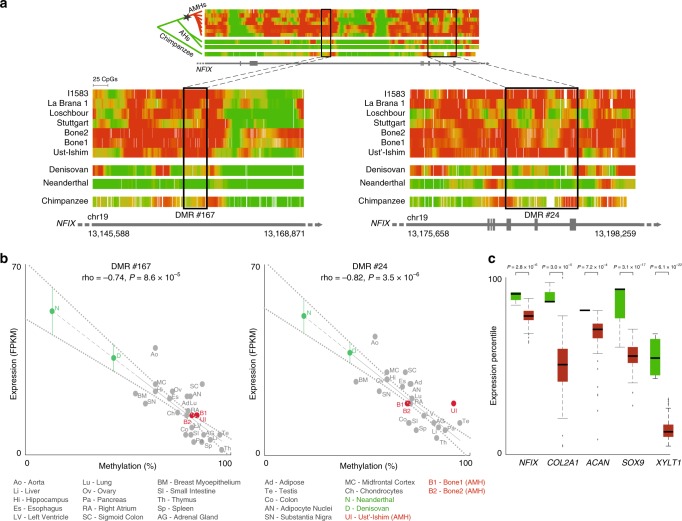
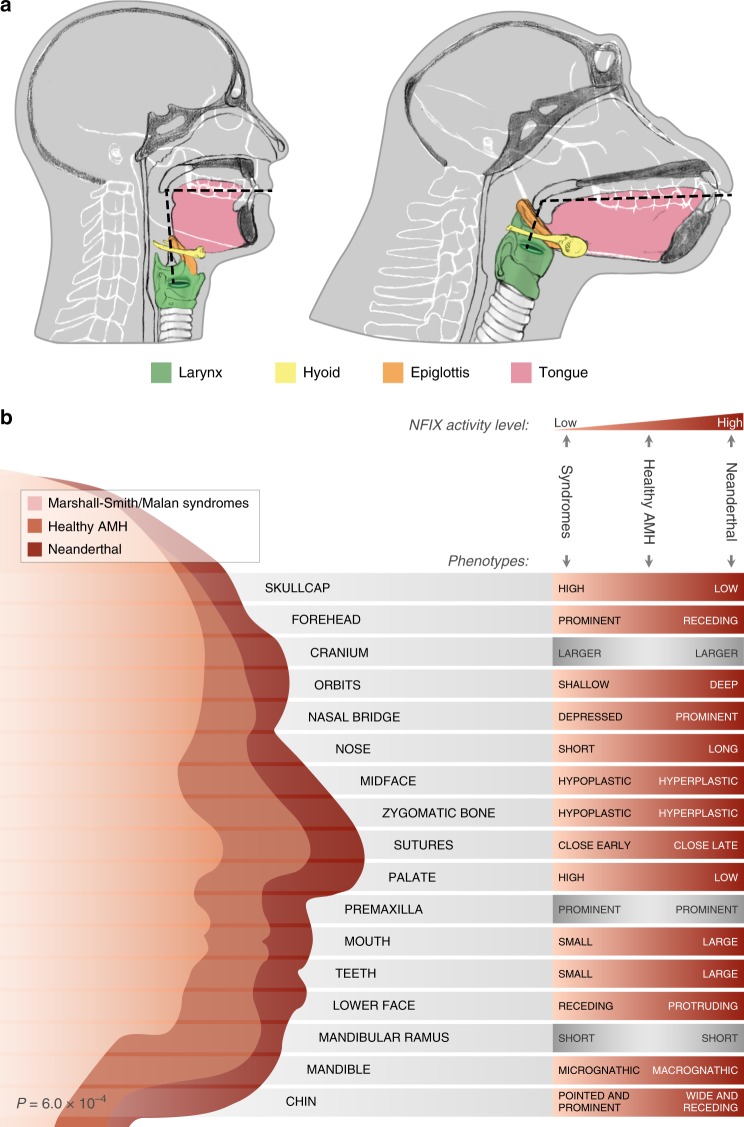
Changes in potential regulatory elements are thought to be key drivers of phenotypic divergence. However, identifying changes to regulatory elements that underlie human-specific traits has proven very challenging. Here, we use 63 reconstructed and experimentally measured DNA methylation maps of ancient and present-day humans, as well as of six chimpanzees, to detect differentially methylated regions that likely emerged in modern humans after the split from Neanderthals and Denisovans. We show that genes associated with face and vocal tract anatomy went through particularly extensive methylation changes. Specifically, we identify widespread hypermethylation in a network of face- and voice-associated genes (SOX9, ACAN, COL2A1, NFIX and XYLT1). We propose that these repression patterns appeared after the split from Neanderthals and Denisovans, and that they might have played a key role in shaping the modern human face and vocal tract.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Gokhman, D. et al. Reconstructing the DNA methylation maps of the Neandertal and the Denisovan. Science344, 523–527 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
