

Disruption of the kringle 1 domain of prothrombin leads to late onset mortality in zebrafish
- PMID: 32132579
- PMCID: PMC7055286
- DOI: 10.1038/s41598-020-60840-7
Disruption of the kringle 1 domain of prothrombin leads to late onset mortality in zebrafish
Abstract
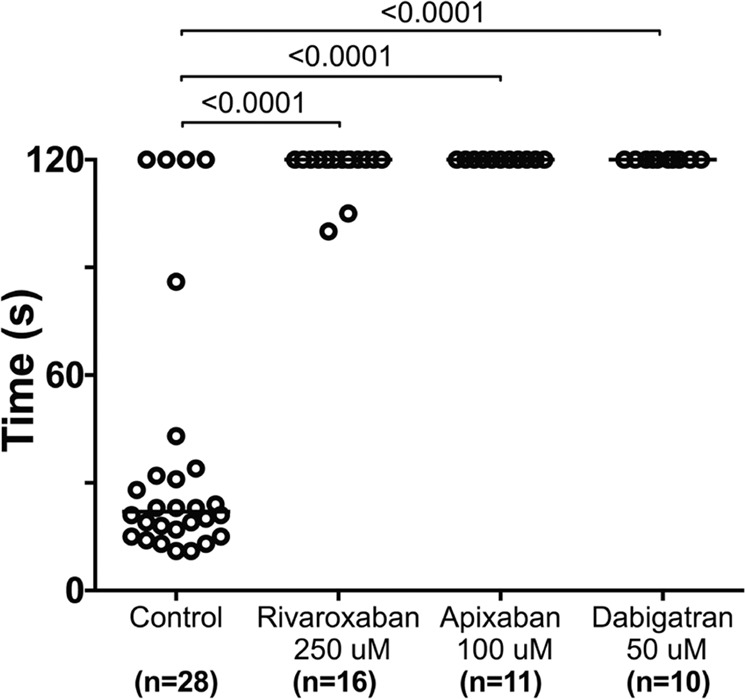
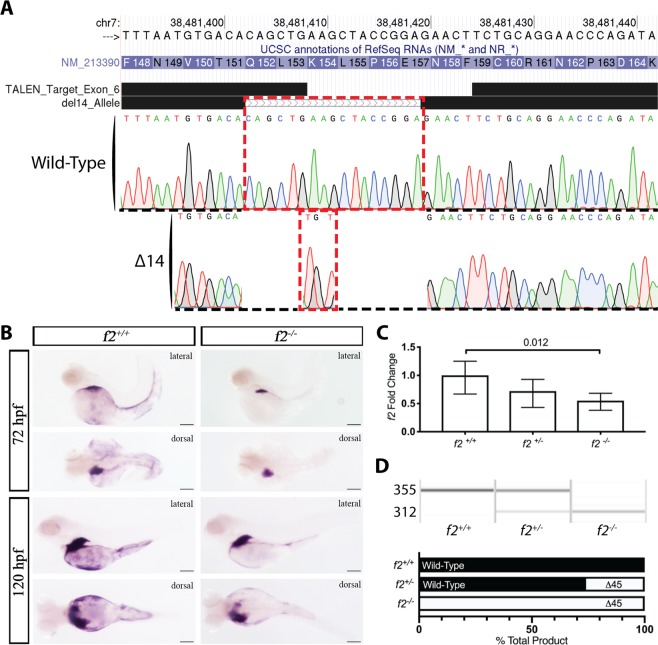
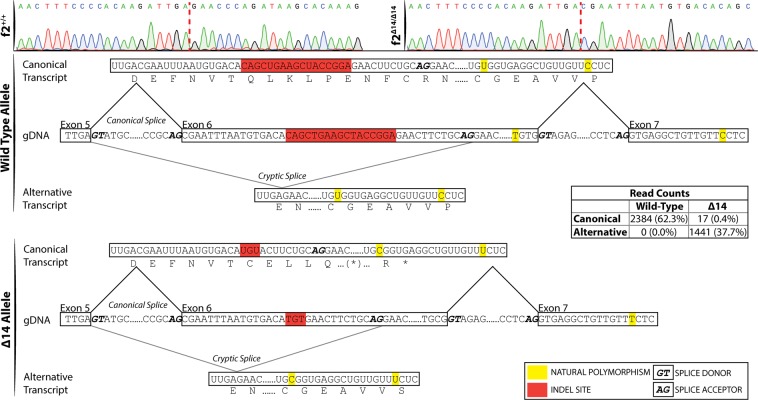
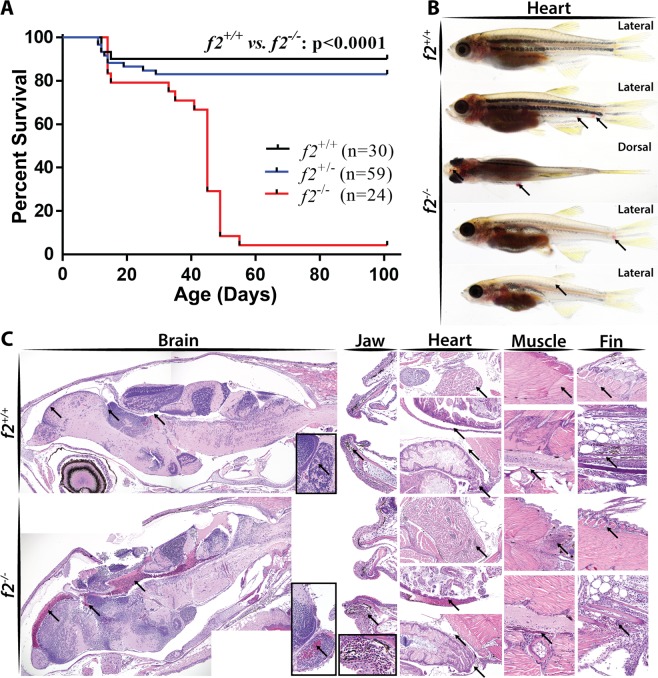
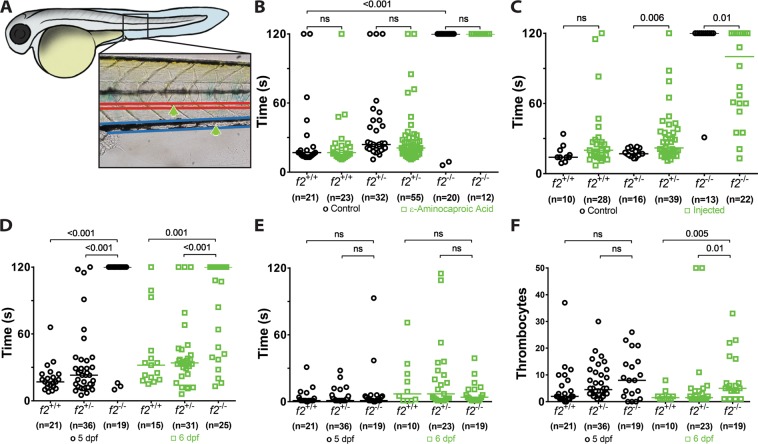
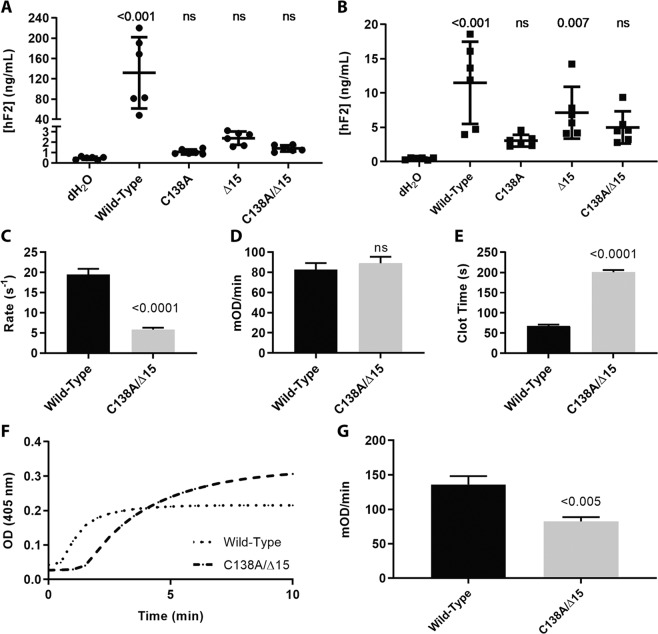
The ability to prevent blood loss in response to injury is a conserved function of all vertebrates. Complete deficiency of the central clotting enzyme prothrombin has never been observed in humans and is incompatible with postnatal life in mice, thus limiting the ability to study its role in vivo. Zebrafish are able to tolerate severe hemostatic deficiencies that are lethal in mammals. We have generated a targeted genetic deletion in the kringle 1 domain of zebrafish prothrombin. Homozygous mutant embryos develop normally into the mid-juvenile stage but demonstrate complete mortality by 2 months of age primarily due to internal hemorrhage. Mutants are unable to form occlusive venous and arterial thrombi in response to endothelial injury, a defect that was phenocopied using direct oral anticoagulants. Human prothrombin engineered with the equivalent mutation exhibits a severe reduction in secretion, thrombin generation, and fibrinogen cleavage. Together, these data demonstrate the conserved function of thrombin in zebrafish and provide insight into the role of kringle 1 in prothrombin maturation and activity. Understanding how zebrafish are able to develop normally and survive into early adulthood without thrombin activity will provide important insight into its pleiotropic functions as well as the management of patients with bleeding disorders.
Conflict of interest statement
J.A.S. has been a consultant for Bayer, Shire, CSL Behring, Spark Therapeutics, and NovoNordisk. J.K.J. has financial interests in Beam Therapeutics, Editas Medicine, Excelsior Genomics, Pairwise Plants, Poseida Therapeutics, Transposagen Biopharmaceuticals, and Verve Therapeutics (f/k/a Endcadia). J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.K.J. is a member of the Board of Directors of the American Society of Gene and Cell Therapy. J.K.J. is a co-inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies. Additional authors have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Figures

References
-
- Rosendaal, F. R. et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb. Haemost. (1998). - PubMed
-
- Poort, S. R., Rosendaal, F. R., Reitsma, P. H. & Bertina, R. M. A Common Genetic Variation in the 3′-Untranslated Region of the Prothrombin Gene Is Associated With Elevated Plasma Prothrombin Levels and an Increase in Venous Thrombosis. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
