

DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR
- PMID: 32133046
- PMCID: PMC7041937
- DOI: 10.18632/oncotarget.27487
DNA dependent protein kinase (DNA-PK) enhances HIV transcription by promoting RNA polymerase II activity and recruitment of transcription machinery at HIV LTR
Abstract
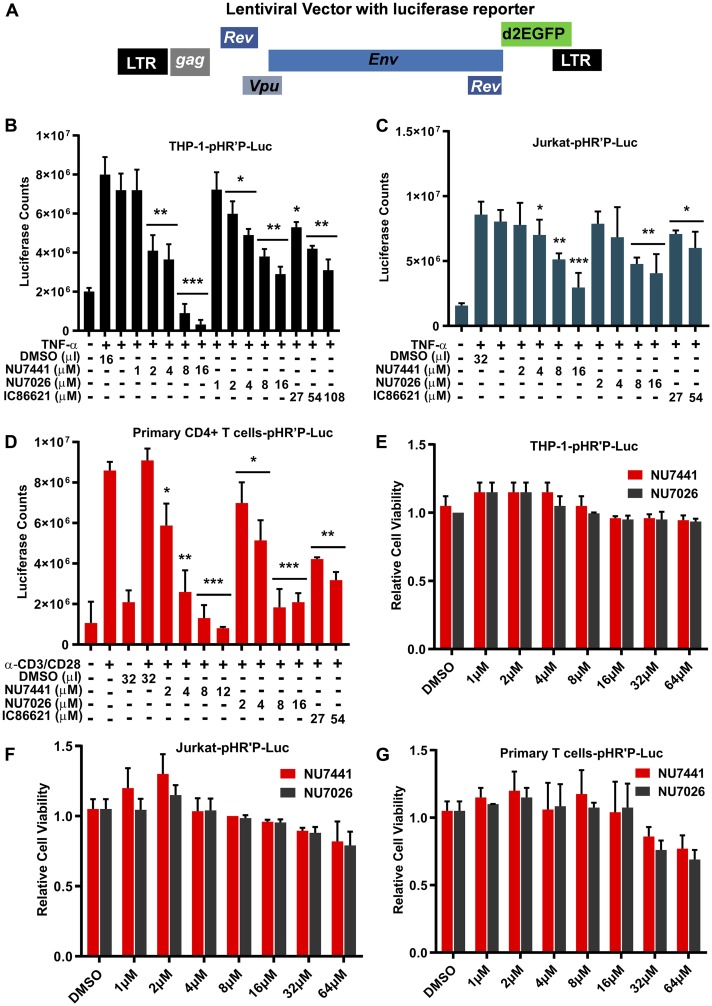
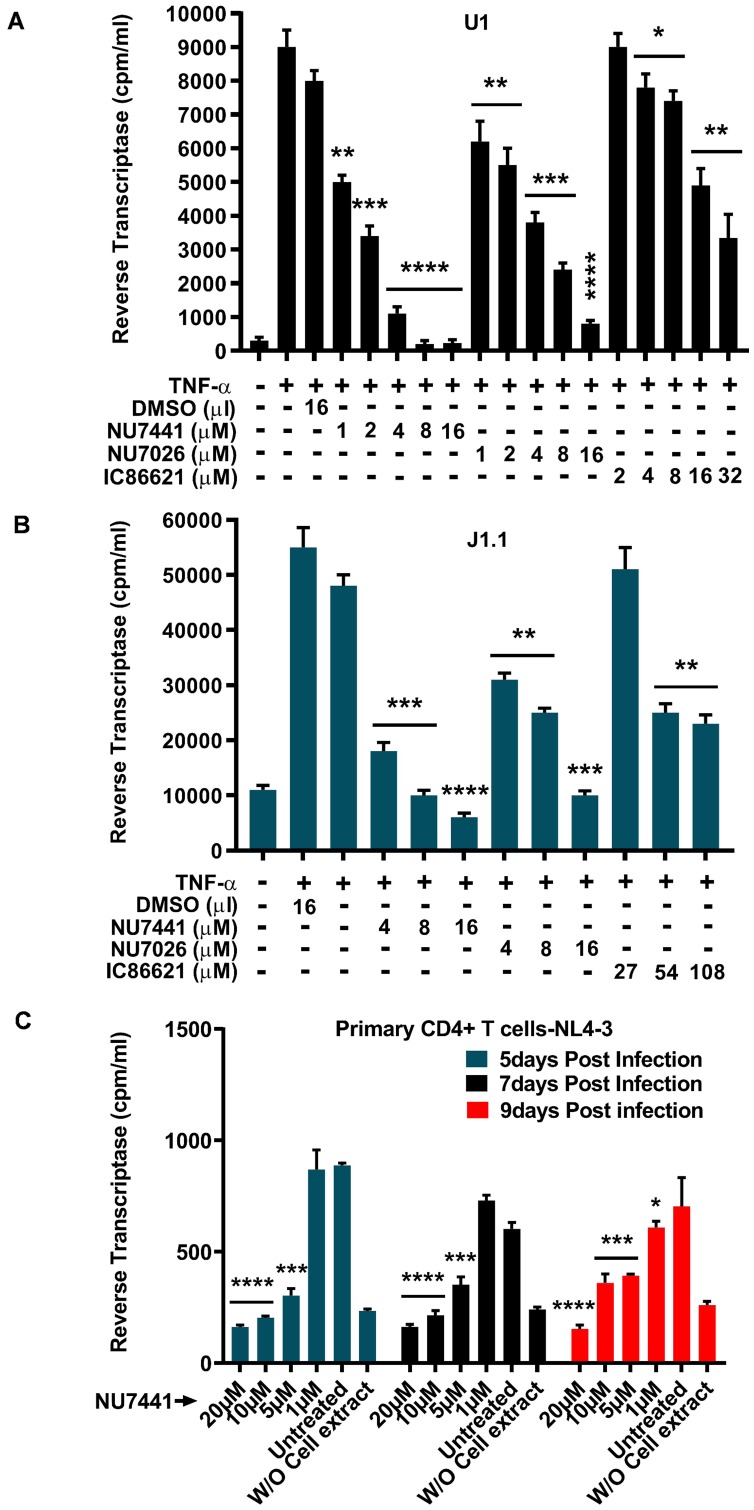
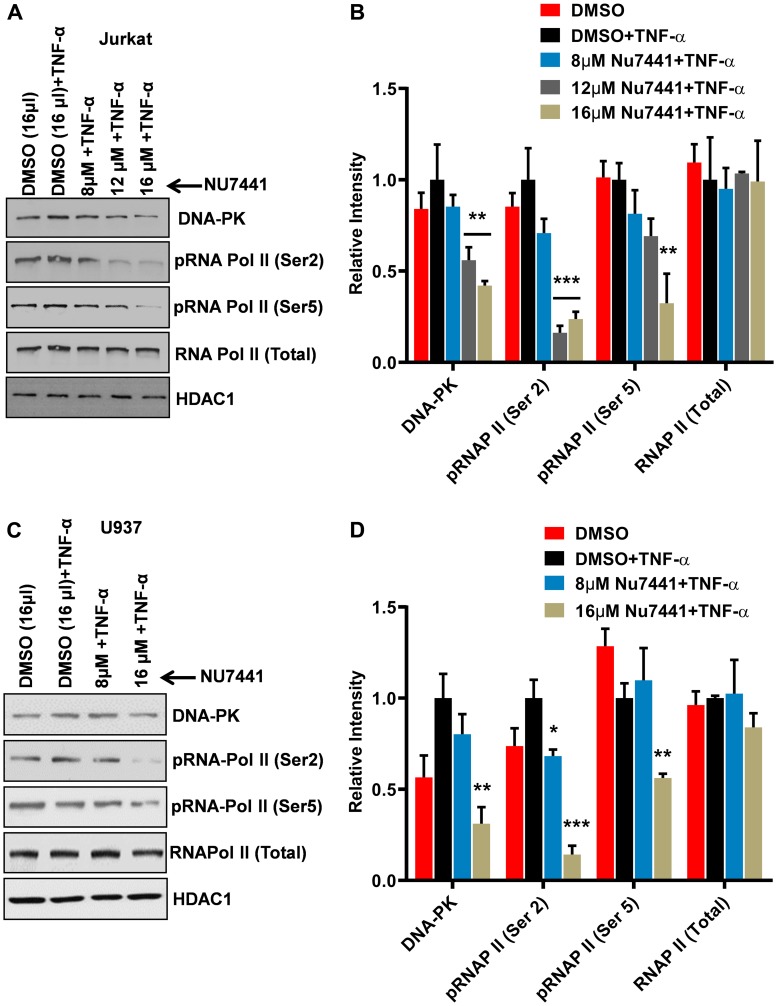
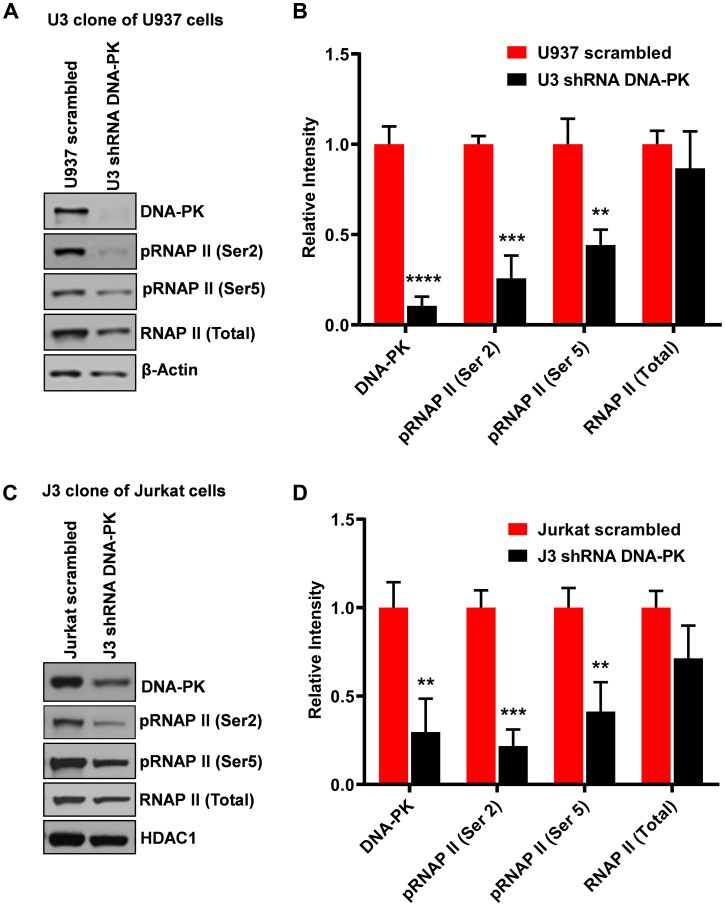
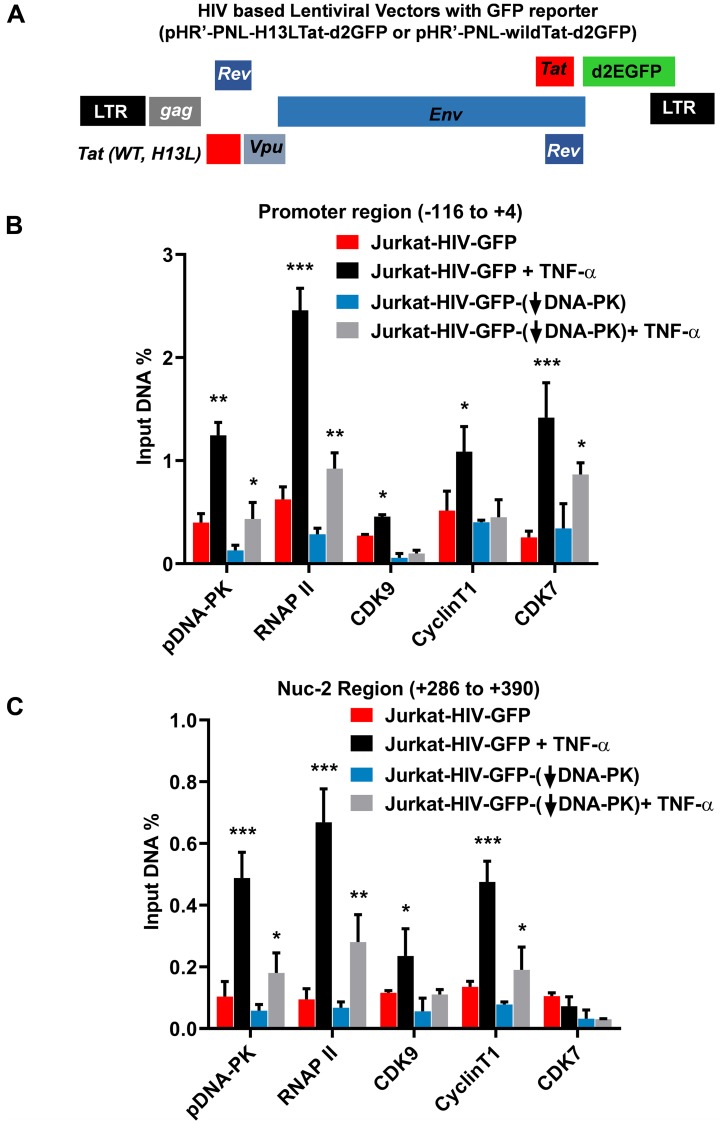
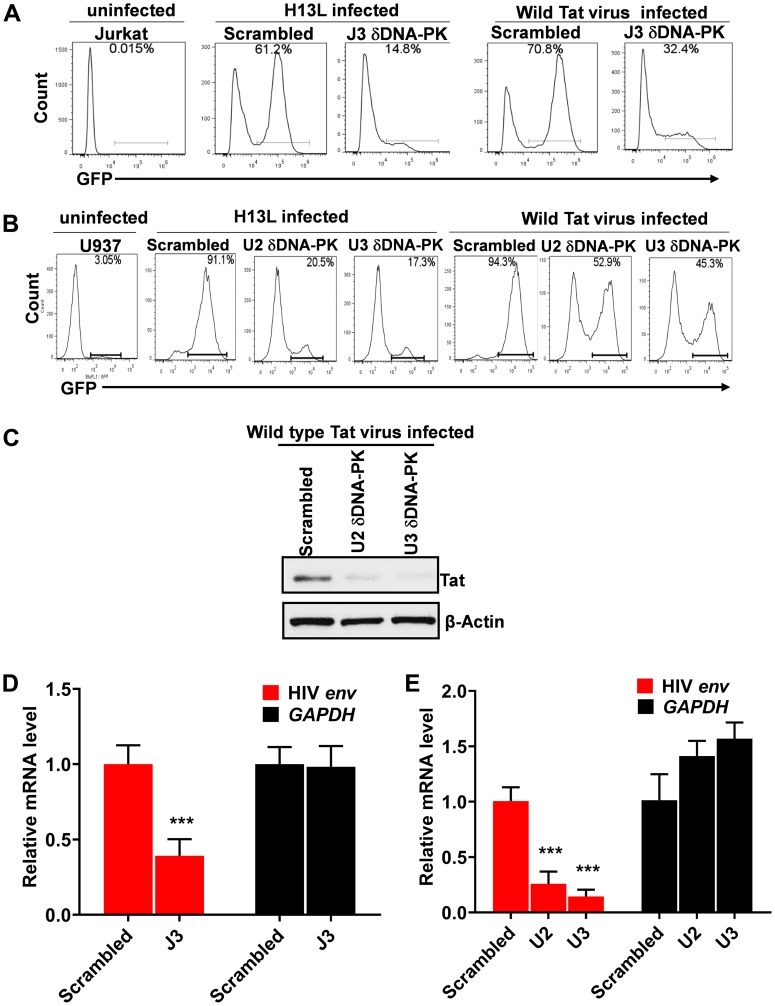
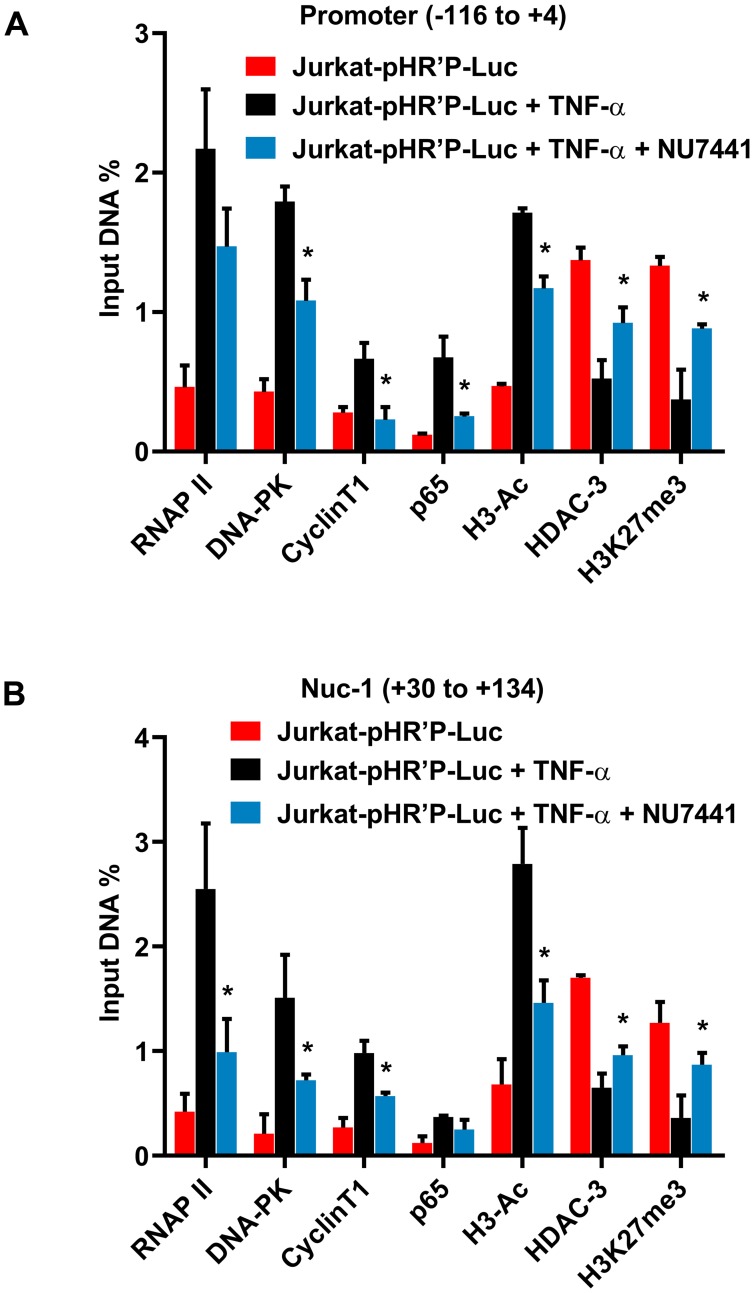
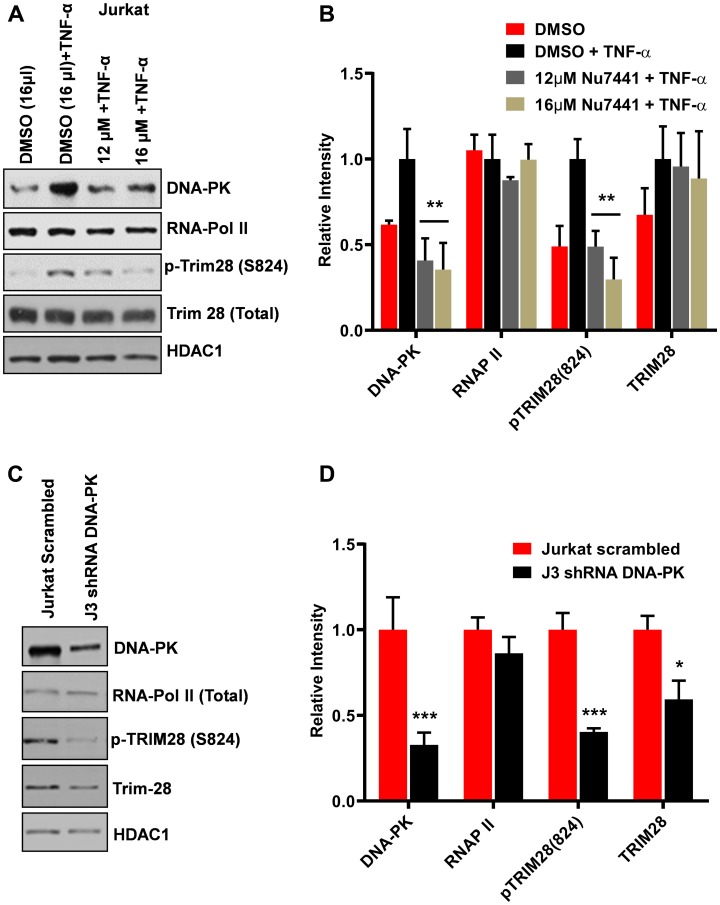
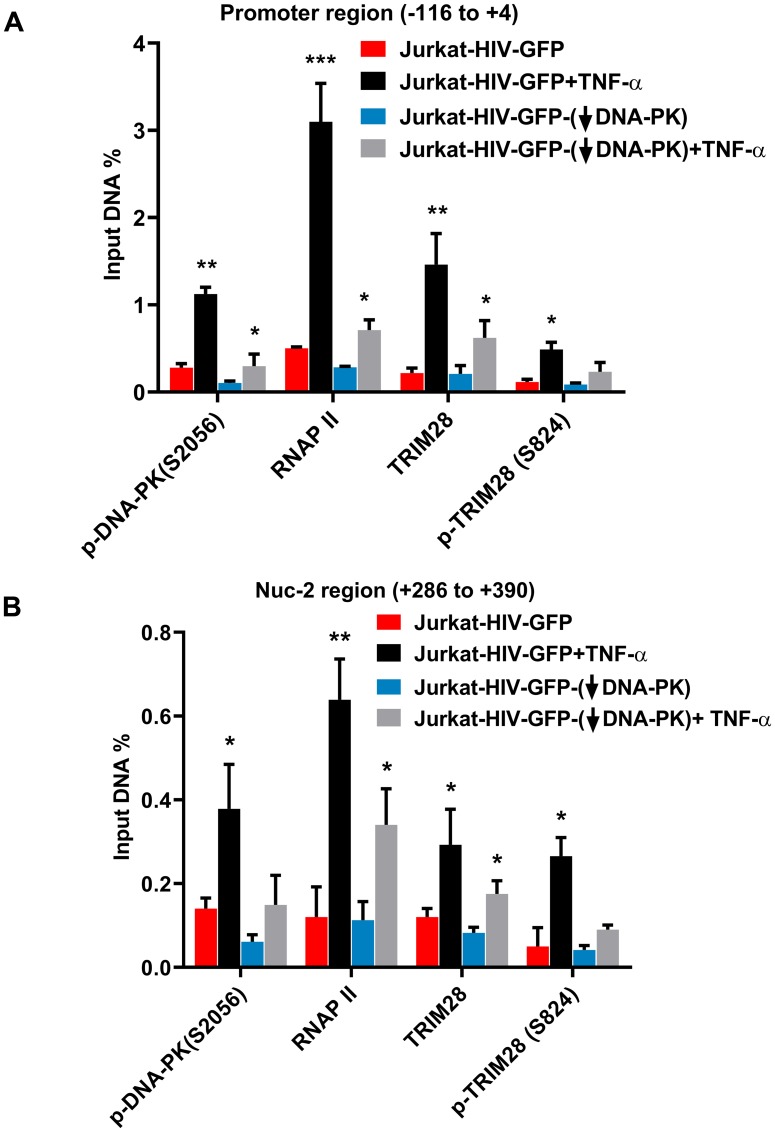
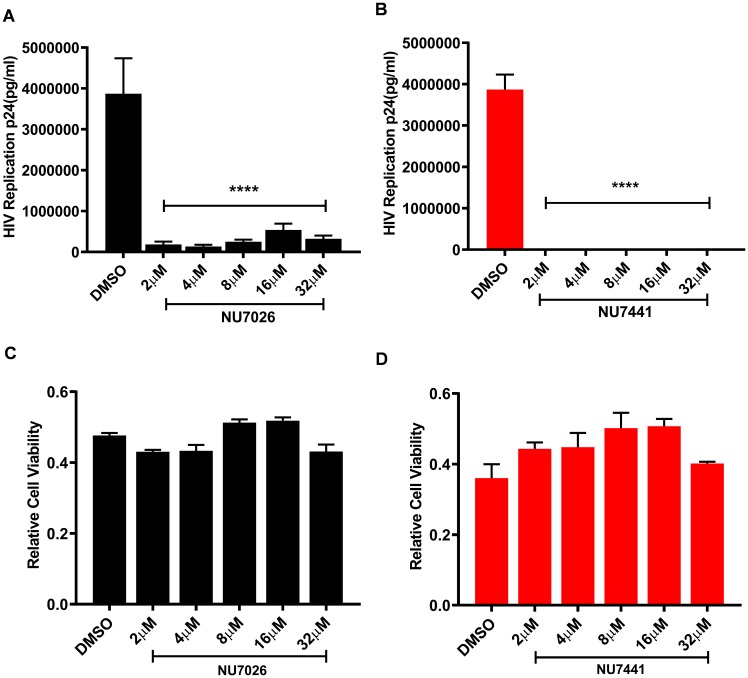
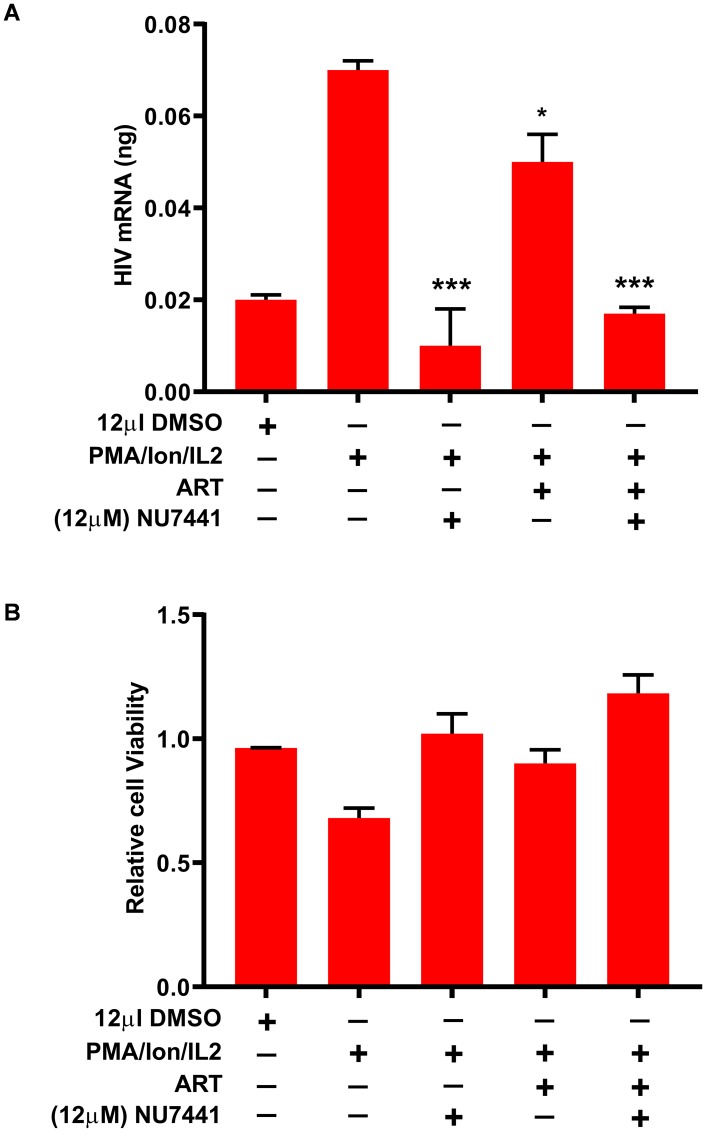
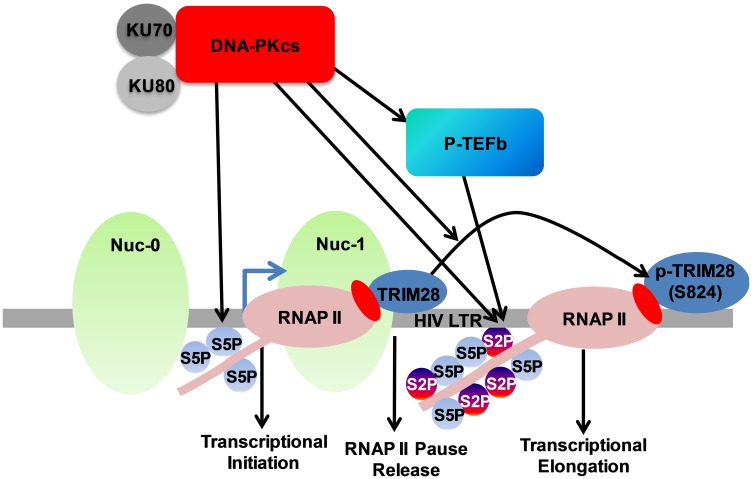
Despite reductions in mortality from the use of highly active antiretroviral therapy (HAART), the presence of latent or transcriptionally silent proviruses prevents HIV cure/eradication. We have previously reported that DNA-dependent protein kinase (DNA-PK) facilitates HIV transcription by interacting with the RNA polymerase II (RNAP II) complex recruited at HIV LTR. In this study, using different cell lines and peripheral blood mononuclear cells (PBMCs) of HIV-infected patients, we found that DNA-PK stimulates HIV transcription at several stages, including initiation, pause-release and elongation. We are reporting for the first time that DNA-PK increases phosphorylation of RNAP II C-terminal domain (CTD) at serine 5 (Ser5) and serine 2 (Ser2) by directly catalyzing phosphorylation and by augmenting the recruitment of the positive transcription elongation factor (P-TEFb) at HIV LTR. Our findings suggest that DNA-PK expedites the establishment of euchromatin structure at HIV LTR. DNA-PK inhibition/knockdown leads to the severe impairment of HIV replication and reactivation of latent HIV provirus. DNA-PK promotes the recruitment of Tripartite motif-containing 28 (TRIM28) at LTR and assists the release of paused RNAP II through TRIM28 phosphorylation. These results provide the mechanisms through which DNA-PK controls the HIV gene expression and, likely, can be extended to cellular gene expression, including during cell malignancy, where the role of DNA-PK has been well-established.
Keywords: DNA-PK; DNA-PK inhibitors; HIV; replication; transcription.
Conflict of interest statement
CONFLICTS OF INTEREST The authors declare that they have no conflicts of interests.
Figures
Similar articles
-
Cocaine-Induced DNA-Dependent Protein Kinase Relieves RNAP II Pausing by Promoting TRIM28 Phosphorylation and RNAP II Hyperphosphorylation to Enhance HIV Transcription.Cells. 2024 Nov 23;13(23):1950. doi: 10.3390/cells13231950. Cells. 2024. PMID: 39682697 Free PMC article.
-
Cocaine-induced DNA-PK relieves RNAP II pausing by promoting TRIM28 phosphorylation.bioRxiv [Preprint]. 2024 Aug 19:2024.08.19.608673. doi: 10.1101/2024.08.19.608673. bioRxiv. 2024. Update in: Cells. 2024 Nov 23;13(23):1950. doi: 10.3390/cells13231950. PMID: 39229050 Free PMC article. Updated. Preprint.
-
Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program.Genes Dev. 2006 Mar 1;20(5):601-12. doi: 10.1101/gad.1398206. Genes Dev. 2006. PMID: 16510875 Free PMC article.
-
The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE.Mol Biosyst. 2017 Jan 31;13(2):246-276. doi: 10.1039/c6mb00387g. Mol Biosyst. 2017. PMID: 27833949 Review.
-
Phosphorylation of the C-terminal domain of RNA polymerase II.Biochim Biophys Acta. 1995 Apr 4;1261(2):171-82. doi: 10.1016/0167-4781(94)00233-s. Biochim Biophys Acta. 1995. PMID: 7711060 Review.
Cited by
-
Crossroads of Drug Abuse and HIV Infection: Neurotoxicity and CNS Reservoir.Vaccines (Basel). 2022 Jan 27;10(2):202. doi: 10.3390/vaccines10020202. Vaccines (Basel). 2022. PMID: 35214661 Free PMC article. Review.
-
Comparison of the Biological Basis for Non-HIV Transmission to HIV-Exposed Seronegative Individuals, Disease Non-Progression in HIV Long-Term Non-Progressors and Elite Controllers.Viruses. 2023 Jun 13;15(6):1362. doi: 10.3390/v15061362. Viruses. 2023. PMID: 37376660 Free PMC article. Review.
-
KAP1/TRIM28: Transcriptional Activator and/or Repressor of Viral and Cellular Programs?Front Cell Infect Microbiol. 2022 Feb 23;12:834636. doi: 10.3389/fcimb.2022.834636. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35281453 Free PMC article. Review.
-
DNA-PKcs: A Targetable Protumorigenic Protein Kinase.Cancer Res. 2022 Feb 15;82(4):523-533. doi: 10.1158/0008-5472.CAN-21-1756. Cancer Res. 2022. PMID: 34893509 Free PMC article. Review.
-
Medicinal chemistry breakthroughs on ATM, ATR, and DNA-PK inhibitors as prospective cancer therapeutics.J Enzyme Inhib Med Chem. 2025 Dec;40(1):2489720. doi: 10.1080/14756366.2025.2489720. Epub 2025 Apr 21. J Enzyme Inhib Med Chem. 2025. PMID: 40256842 Free PMC article. Review.
References
-
- Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, et al.. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A. 1999; 96:15109–15114. 10.1073/pnas.96.26.15109. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
