

In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery
- PMID: 32133344
- PMCID: PMC7040036
- DOI: 10.3389/fchem.2020.00093
In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery
Abstract
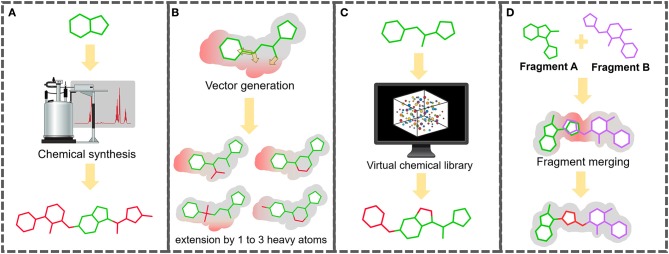
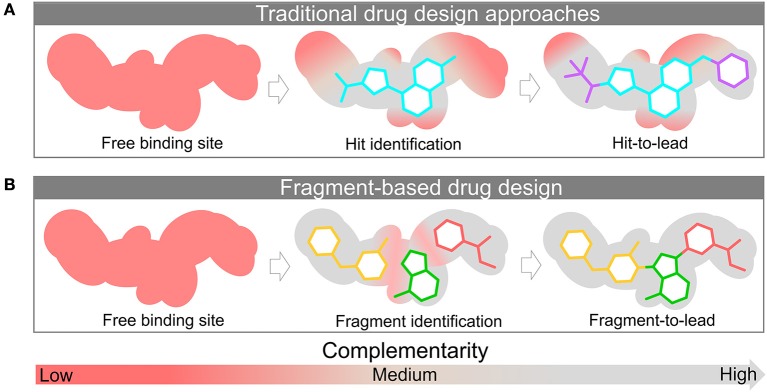
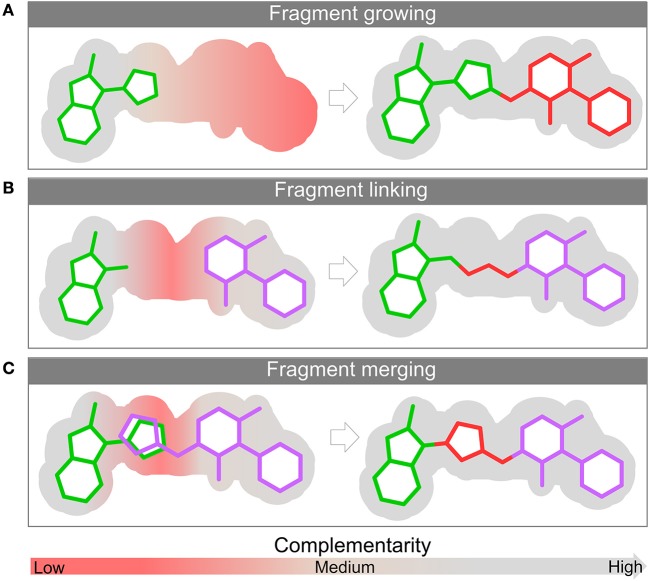
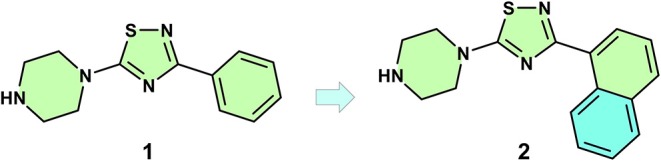
Fragment-based drug (or lead) discovery (FBDD or FBLD) has developed in the last two decades to become a successful key technology in the pharmaceutical industry for early stage drug discovery and development. The FBDD strategy consists of screening low molecular weight compounds against macromolecular targets (usually proteins) of clinical relevance. These small molecular fragments can bind at one or more sites on the target and act as starting points for the development of lead compounds. In developing the fragments attractive features that can translate into compounds with favorable physical, pharmacokinetics and toxicity (ADMET-absorption, distribution, metabolism, excretion, and toxicity) properties can be integrated. Structure-enabled fragment screening campaigns use a combination of screening by a range of biophysical techniques, such as differential scanning fluorimetry, surface plasmon resonance, and thermophoresis, followed by structural characterization of fragment binding using NMR or X-ray crystallography. Structural characterization is also used in subsequent analysis for growing fragments of selected screening hits. The latest iteration of the FBDD workflow employs a high-throughput methodology of massively parallel screening by X-ray crystallography of individually soaked fragments. In this review we will outline the FBDD strategies and explore a variety of in silico approaches to support the follow-up fragment-to-lead optimization of either: growing, linking, and merging. These fragment expansion strategies include hot spot analysis, druggability prediction, SAR (structure-activity relationships) by catalog methods, application of machine learning/deep learning models for virtual screening and several de novo design methods for proposing synthesizable new compounds. Finally, we will highlight recent case studies in fragment-based drug discovery where in silico methods have successfully contributed to the development of lead compounds.
Keywords: de novo design; drug discovery; fragment-based; hot spot analysis; in silico methods; lead discovery; machine learning; optimization.
Copyright © 2020 de Souza Neto, Moreira-Filho, Neves, Maidana, Guimarães, Furnham, Andrade and Silva.
Figures
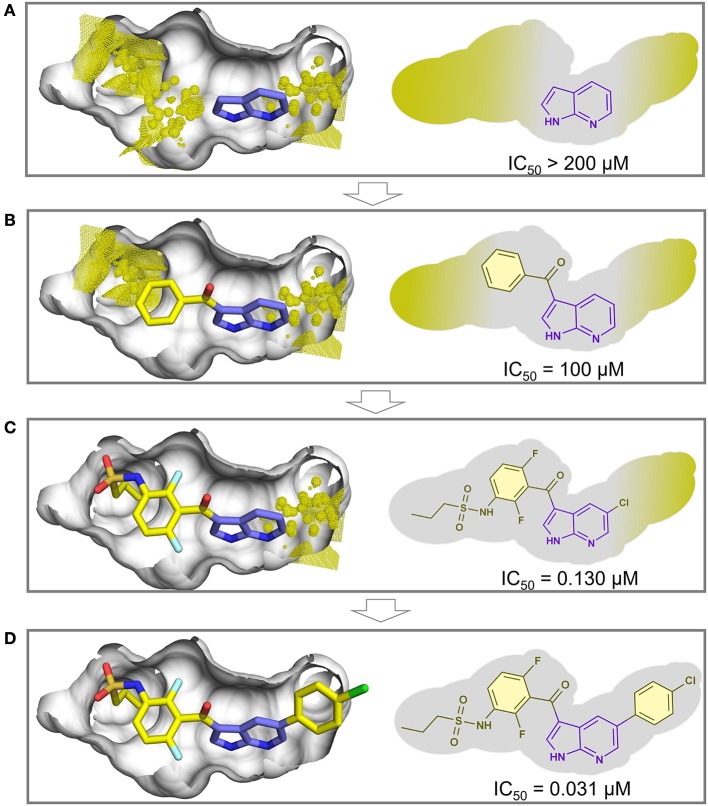
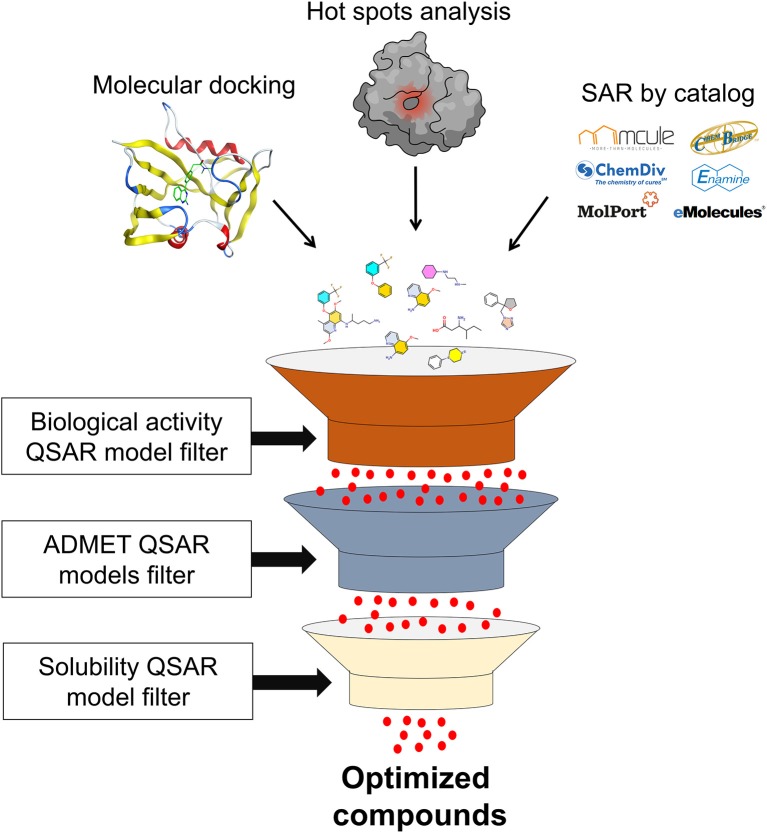
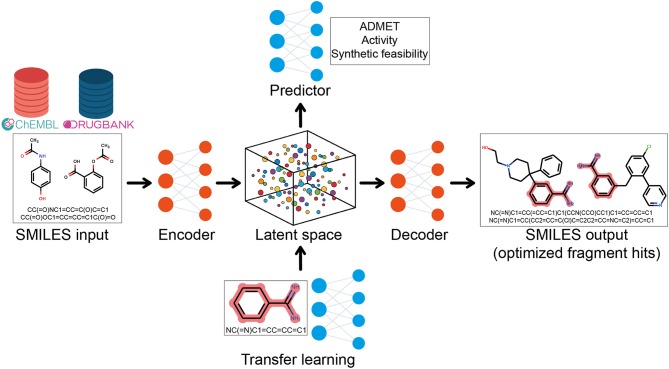
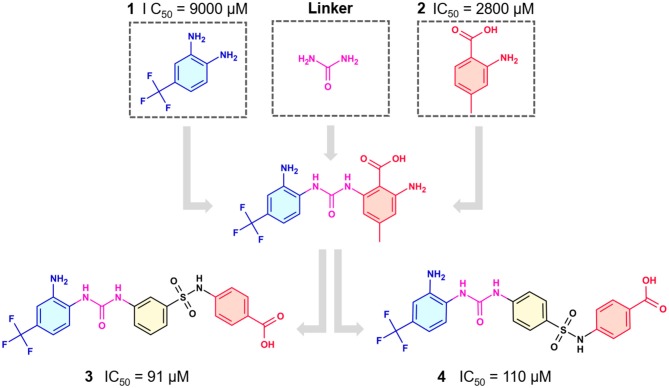
References
-
- Abad-Zapatero C. (2013). Ligand Efficiency Indices for Drug Discovery. Oxford, UK: Elsevier.
-
- Alves V., Braga R., Muratov E., Andrade C. (2018). Development of web and mobile applications for chemical toxicity prediction. J. Braz. Chem. Soc. 29, 982–988. 10.21577/0103-5053.20180013 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous