

Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models
- PMID: 32133356
- PMCID: PMC7039819
- DOI: 10.3389/fcell.2020.00027
Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models
Abstract
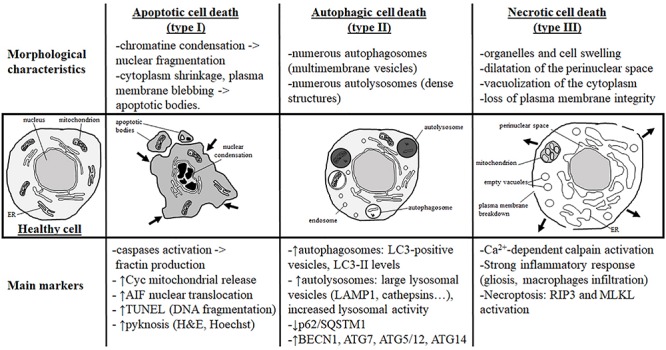
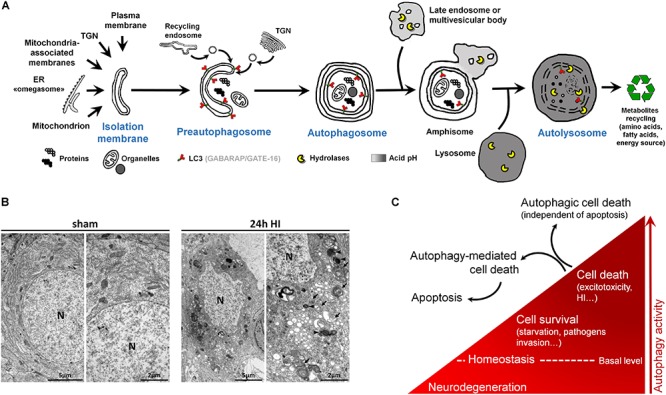
Despite tremendous advances in neonatal intensive care over the past 20 years, prematurity carries a high burden of neurological morbidity lasting lifelong. The term encephalopathy of prematurity (EoP) coined by Volpe in 2009 encompasses all aspects of the now known effects of prematurity on the immature brain, including altered and disturbed development as well as specific lesional hallmarks. Understanding the way cells are damaged is crucial to design brain protective strategies, and in this purpose, preclinical models largely contribute to improve the comprehension of the cell death mechanisms. While neuronal cell death has been deeply investigated and characterized in (hypoxic-ischemic) encephalopathy of the newborn at term, little is known about the types of cell death occurring in preterm brain injury. Three main different morphological cell death types are observed in the immature brain, specifically in models of hypoxic-ischemic encephalopathy, namely, necrotic, apoptotic, and autophagic cell death. Features of all three types may be present in the same dying neuron. In preterm brain injury, description of cell death types is sparse, and cell loss primarily concerns immature oligodendrocytes and, infrequently, neurons. In the present review, we first shortly discuss the different main severe preterm brain injury conditions that have been reported to involve cell death, including periventricular leucomalacia (PVL), diffuse white matter injury (dWMI), and intraventricular hemorrhages, as well as potentially harmful iatrogenic conditions linked to premature birth (anesthesia and caffeine therapy). Then, we present an overview of current evidence concerning cell death in both clinical human tissue data and preclinical models by focusing on studies investigating the presence of cell death allowing discriminating between the types of cell death involved. We conclude that, to improve brain protective strategies, not only apoptosis but also other cell death (such as regulated necrotic and autophagic) pathways now need to be investigated together in order to consider all cell death mechanisms involved in the pathogenesis of preterm brain damage.
Keywords: apoptosis; autophagic cell death; autophagy; necrosis; neonatal; neuroprotection; periventricular leucomalacia.
Copyright © 2020 Truttmann, Ginet and Puyal.
Figures
References
-
- Alonso-Alconada D., Alvarez F. J., Alvarez A., Mielgo V. E., Goni-de-Cerio F., Rey-Santano M. C., et al. (2010). The cannabinoid receptor agonist WIN 55,212-2 reduces the initial cerebral damage after hypoxic-ischemic injury in fetal lambs. Brain Res. 1362 150–159. 10.1016/j.brainres.2010.09.050 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous