

Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes
- PMID: 32133419
- PMCID: PMC7039900
- DOI: 10.1038/s41698-020-0109-y
Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes
Abstract
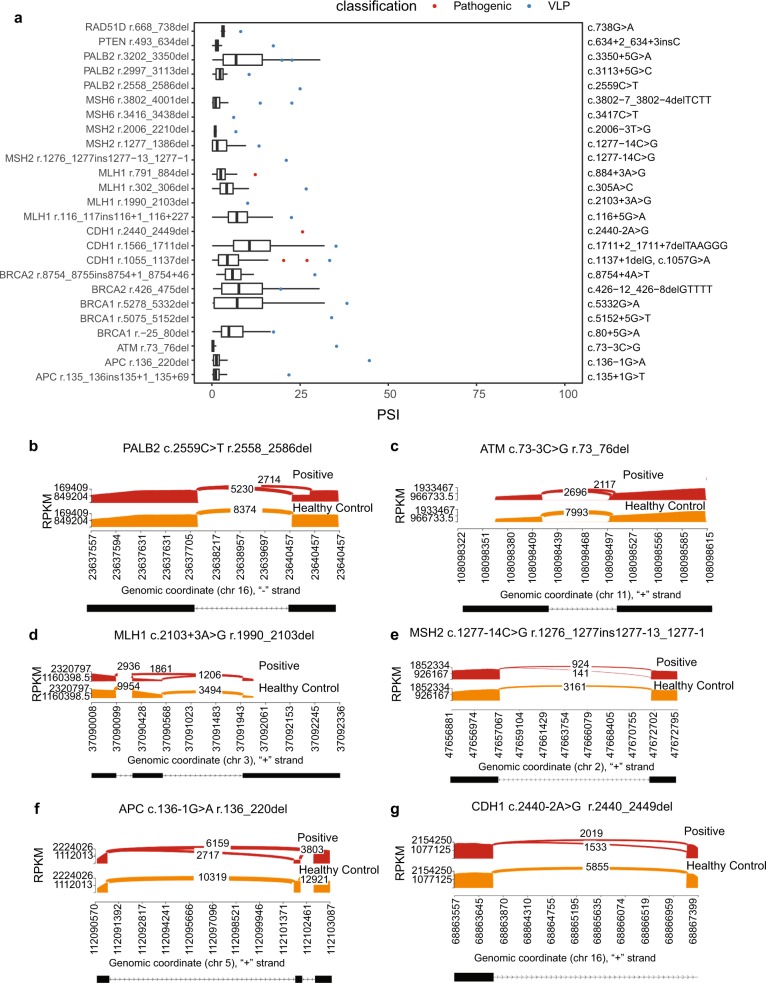
Germline variants in tumor suppressor genes (TSGs) can result in RNA mis-splicing and predisposition to cancer. However, identification of variants that impact splicing remains a challenge, contributing to a substantial proportion of patients with suspected hereditary cancer syndromes remaining without a molecular diagnosis. To address this, we used capture RNA-sequencing (RNA-seq) to generate a splicing profile of 18 TSGs (APC, ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NF1, PALB2, PMS2, PTEN, RAD51C, RAD51D, and TP53) in 345 whole-blood samples from healthy donors. We subsequently demonstrated that this approach can detect mis-splicing by comparing splicing profiles from the control dataset to profiles generated from whole blood of individuals previously identified with pathogenic germline splicing variants in these genes. To assess the utility of our TSG splicing profile to prospectively identify pathogenic splicing variants, we performed concurrent capture DNA and RNA-seq in a cohort of 1000 patients with suspected hereditary cancer syndromes. This approach improved the diagnostic yield in this cohort, resulting in a 9.1% relative increase in the detection of pathogenic variants, demonstrating the utility of performing simultaneous DNA and RNA genetic testing in a clinical context.
Keywords: Cancer genetics; Genetic testing; Next-generation sequencing.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsT.L., B.L., A.C., B.C., H.L., S.W., H.V., D.X., A.A., M.P., L.H., B.M., M.F., S.F., S.C., J.B., T.P., J.P., B.S., G.H., E.D., J.R.-R.C., R.B., H.-M.L., B.T.-D., A.E., E.C., and R.K. were employees of Ambry Genetics during the time this study was conducted. All other authors have nothing to disclose.
Figures



References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
