

Thermal proteome profiling for interrogating protein interactions
- PMID: 32133759
- PMCID: PMC7057112
- DOI: 10.15252/msb.20199232
Thermal proteome profiling for interrogating protein interactions
Abstract
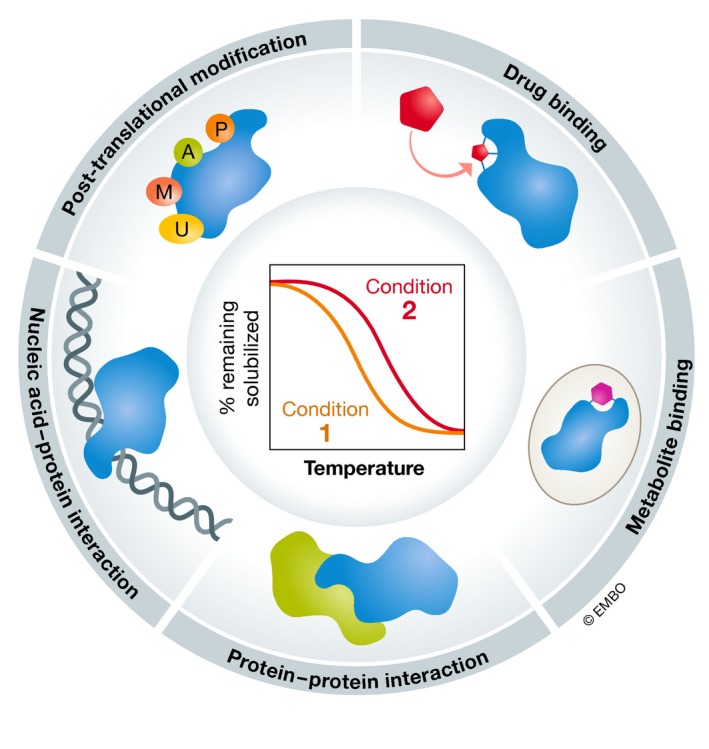
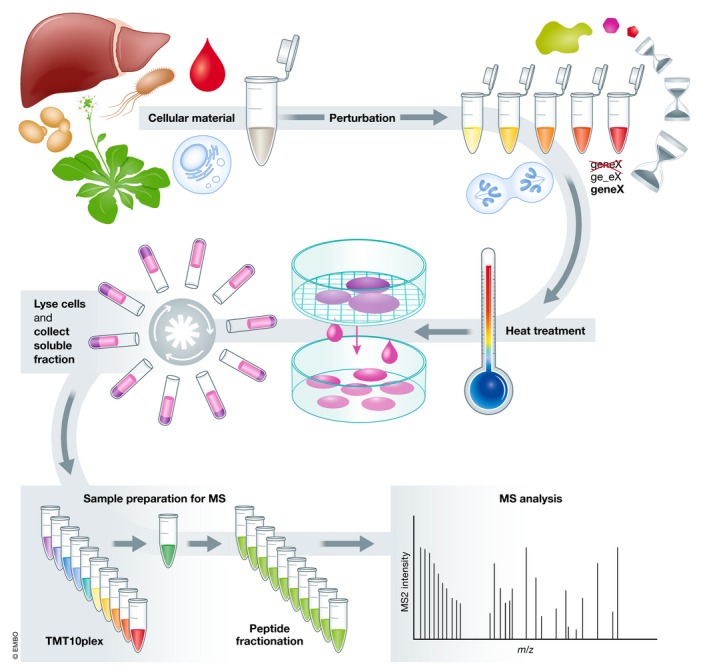
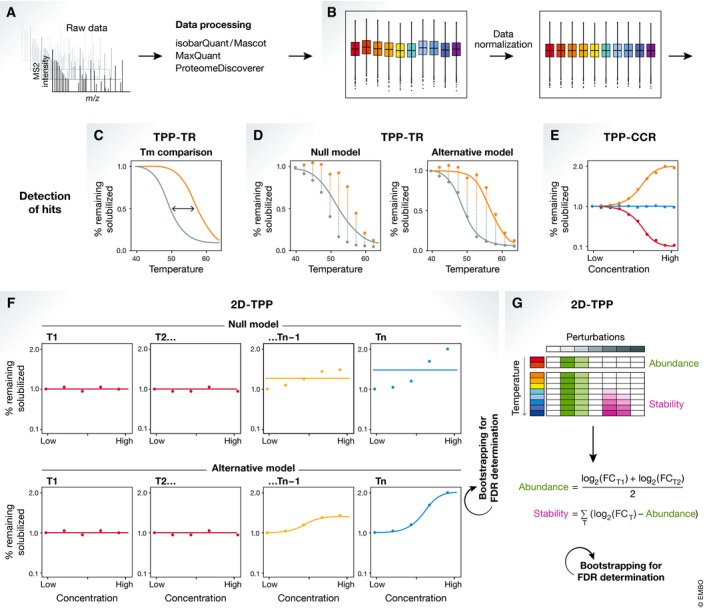
Thermal proteome profiling (TPP) is based on the principle that, when subjected to heat, proteins denature and become insoluble. Proteins can change their thermal stability upon interactions with small molecules (such as drugs or metabolites), nucleic acids or other proteins, or upon post-translational modifications. TPP uses multiplexed quantitative mass spectrometry-based proteomics to monitor the melting profile of thousands of expressed proteins. Importantly, this approach can be performed in vitro, in situ, or in vivo. It has been successfully applied to identify targets and off-targets of drugs, or to study protein-metabolite and protein-protein interactions. Therefore, TPP provides a unique insight into protein state and interactions in their native context and at a proteome-wide level, allowing to study basic biological processes and their underlying mechanisms.
Keywords: drug discovery; metabolites; protein complexes; proteomics; thermal proteome profiling.
© 2020 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
