

Designing Novel Therapies to Mend Broken Hearts: ATF6 and Cardiac Proteostasis
- PMID: 32138230
- PMCID: PMC7140506
- DOI: 10.3390/cells9030602
Designing Novel Therapies to Mend Broken Hearts: ATF6 and Cardiac Proteostasis
Abstract
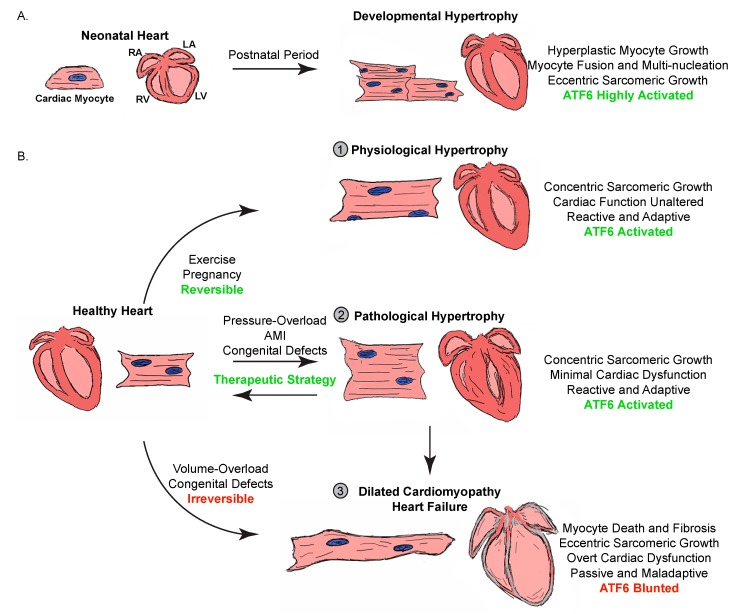
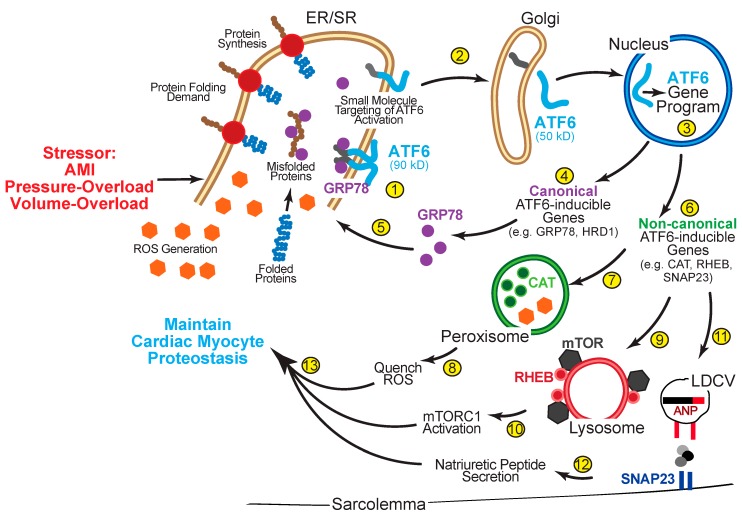
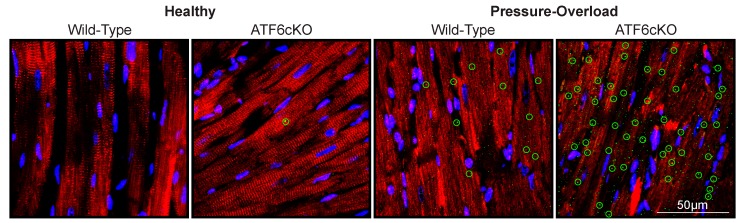
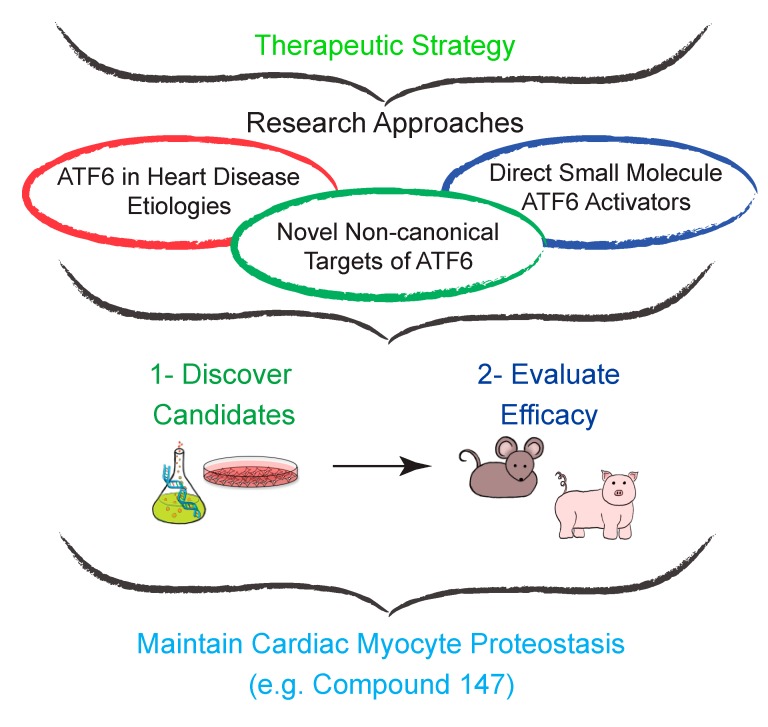
The heart exhibits incredible plasticity in response to both environmental and genetic alterations that affect workload. Over the course of development, or in response to physiological or pathological stimuli, the heart responds to fluctuations in workload by hypertrophic growth primarily by individual cardiac myocytes growing in size. Cardiac hypertrophy is associated with an increase in protein synthesis, which must coordinate with protein folding and degradation to allow for homeostatic growth without affecting the functional integrity of cardiac myocytes (i.e., proteostasis). This increase in the protein folding demand in the growing cardiac myocyte activates the transcription factor, ATF6 (activating transcription factor 6α, an inducer of genes that restore proteostasis. Previously, ATF6 has been shown to induce ER-targeted proteins functioning primarily to enhance ER protein folding and degradation. More recent studies, however, have illuminated adaptive roles for ATF6 functioning outside of the ER by inducing non-canonical targets in a stimulus-specific manner. This unique ability of ATF6 to act as an initial adaptive responder has bolstered an enthusiasm for identifying small molecule activators of ATF6 and similar proteostasis-based therapeutics.
Keywords: ATF6; cardiac myocyte; cardiomyopathy; hypertrophy; proteostasis; small molecule; therapy; transcriptional regulation; unfolded protein response (UPR).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Benjamin E.J., Virani S.S., Callaway C.W., Chamberlain A.M., Chang A.R., Cheng S., Chiuve S.E., Cushman M., Delling F.N., Deo R. American Heart Association Council on Prevention Statistics and Stroke Statistics Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:467–492. doi: 10.1161/CIR.0000000000000558. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
