

Molecular Genetics of Niemann-Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants
- PMID: 32138288
- PMCID: PMC7141276
- DOI: 10.3390/jcm9030679
Molecular Genetics of Niemann-Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants
Abstract
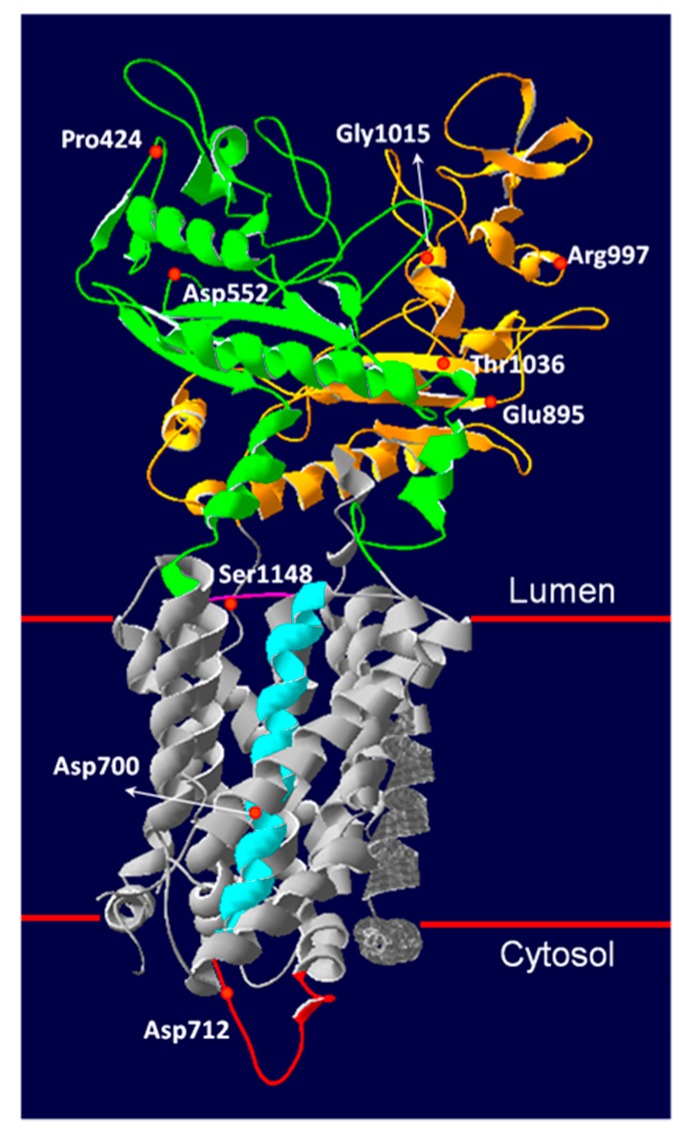
Niemann-Pick type C (NPC) disease is an autosomal recessive lysosomal storage disorder caused by mutations in NPC1 or NPC2 genes. In 2009, the molecular characterization of 44 NPC Italian patients has been published. Here, we present an update of the genetic findings in 105 Italian NPC patients belonging to 83 unrelated families (77 NPC1 and 6 NPC2). NPC1 and NPC2 genes were studied following an algorithm recently published. Eighty-four different NPC1 and five NPC2 alleles were identified. Only two NPC1 alleles remained non detected. Sixty-two percent of NPC1 alleles were due to missense variants. The most frequent NPC1 mutation was the p.F284Lfs*26 (5.8% of the alleles). All NPC2 mutations were found in the homozygous state, and all but one was severe. Among newly diagnosed patients, 18 novel NPC1 mutations were identified. The pathogenic nature of 7/9 missense alleles and 3/4 intronic variants was confirmed by filipin staining and NPC1 protein analysis or mRNA expression in patient's fibroblasts. Taken together, our previous published data and new results provide an overall picture of the molecular characteristics of NPC patients diagnosed so far in Italy.
Keywords: NPC1; NPC2; Niemann–Pick C disease; mutations.
Conflict of interest statement
A.D. has received research grants from Actelion Pharmaceuticals and Orphazyme. C.C. was supported by an educational grant from Actelion Pharmaceuticals. S.Z., C.G., R.C., S.C., P.P., R.D., A.S., D.V., S.G., A.F., S.F., M.C., M.F., L.S., B.B. (Barbara Borroni), A.B., F.B., C.V.R., M.D., A.T., M.S., and B.B. (Bruno Bembi) have no conflict of interests to declare.
Figures





References
-
- Wassif C.A., Cross J.L., Iben J., Sanchez-Pulido L., Cougnoux A., Platt F.M., Ory D.S., Ponting C.P., Bailey-Wilson J.E., Biesecker L.G., et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016;18:41–48. doi: 10.1038/gim.2015.25. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
